In uno studio condotto questa settimana su Science Translational Medicine, un team internazionale guidato da ricercatori del Vanderbilt University Medical Center getta una nuova luce sulla causa della fibrosi polmonare e dimostra un modo per impedire la malattia nei topi.
I risultati dello studio implicano che alcuni farmaci già presenti sul mercato potrebbero essere riproposti per inibire o arrestare questa grave malattia respiratoria.
Wonder Drake, Professore di Medicina e Patologia, Microbiologia e Immunologia ha guidato lo studio con Lindsay Celada, istruttore di ricerca in Medicina.
“Sono doppiamente entusiasta della prospettiva di questi studi clinici che procedono proprio qui alla VUMC: una combinazione di talento investigativo clinico e infrastruttura di ricerca rende VUMC il posto naturale per far avanzare le scoperte del nostro laboratorio nel regno clinico”.
( Vedi anche: Scoperti i meccanismi responsabili dello sviluppo della fibrosi polmonare idiopatica).
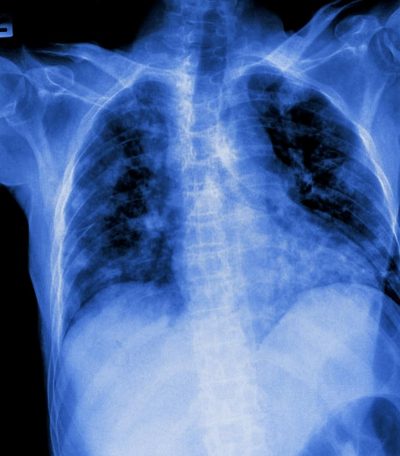
Le persone con fibrosi polmonare sono costantemente a corto di fiato. Il tessuto connettivo nei polmoni si è ispessito o cicatrizzato, riducendo l’apporto di ossigeno al sangue.
La fibrosi si verifica in una serie di malattie che colpiscono i polmoni, tra cui la sarcoidosi e la fibrosi polmonare idiopatica (IPF) e la sopravvivenza media è di circa tre anni. La fibrosi polmonare può anche verificarsi in risposta alla bleomicina, un farmaco in uso dagli anni ’60 per curare il cancro.
Utilizzando risultati precedenti, il nuovo studio rivela un meccanismo immunologico comune alla base di questi tre diversi tipi di malattia.
In entrambi i casi di fibrosi indotta da IPF e bleomicina, studi precedenti avevano indicato connessioni con una proteina di segnalazione proinfiammatoria chiamata interleuchina-17 (IL-17A). Nel frattempo, le persone che studiavano la sarcoidosi avevano trovato una disfunzione in un tipo di globuli bianchi chiamati T CD4 +.
Drake aveva rintracciato questa disfunzione di T CD4 + nella sovraregolazione di PD-1 o proteina programmata per la morte cellulare, un recettore della superficie cellulare che inibisce l’autoimmunità.
I fibroblasti sono cellule che producono la struttura dei tessuti animali, inclusi i tessuti cicatrizzanti. In particolare, è stato dimostrato che IPF coinvolge l’iperattivazione dei fibroblasti polmonari .
Il team ha inserito i fibroblasti polmonari normali in coltura con cellule T CD4 + raccolte da persone con IPF o sarcoidosi. La produzione di collagene di tipo 1, un segno distintivo della fibrosi, è risultata in entrambe queste culture, ma non nel caso di cellule T CD4 + da pazienti con polmoni sani.
Impiegando una serie di tecniche e coprendo molte aree, lo studio introduce una storia causale che inizia con le cellule T CD4 + e sovraregolate PD-1, portando ad una maggiore espressione di un attivatore di trascrizione genica chiamato STAT3 e quindi ad una maggiore produzione di IL- 17A, con conseguente fibrosi.
“Anche se in genere non sono stati considerati importanti nella fibrosi polmonare, stiamo dimostrando un ruolo centrale in una serie di malattie, delle cellule T CD4 + con sovraregolazione PD-1“, ha detto Celada.
Con il suo ampio campo di applicazione, lo studio pone l’attenzione su tre potenziali bersagli farmacologici per i quali sono già disponibili farmaci: il blocco del PD-1 può ridurre significativamente la fibrosi nei topi; utilizzando cellule umane in vitro, i ricercatori hanno dimostrato che l’inibizione chimica di STAT3 riduce i componenti della fibrosi, come la produzione di collagene e infine, lo studio apporta maggiore attenzione al ruolo di IL-17A nella fibrosi polmonare della sarcoidosi.
Fonte: Medicalxpress