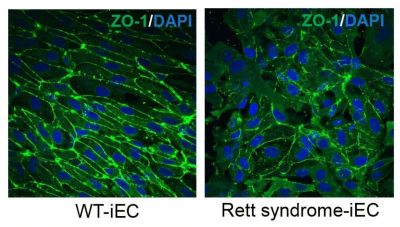
Sindrome di Rett-immagine: in questo dettaglio di una figura tratta dall’articolo, le cellule endoteliali della sindrome di Rett (a destra) mostrano una minore espressione di ZO-1 (verde), una proteina chiave per la formazione di una chiusura ermetica nei vasi sanguigni. Le cellule di controllo (a sinistra) mostrano una ZO-1 molto più completa. Crediti: Molecular Psychiatry (2026).
La sindrome di Rett (RTT) è un disturbo dello sviluppo neurologico causato da mutazioni nella proteina legante metil-CpG 2 (MeCP2). MeCP2 è una proteina legante il DNA non specifica per tipo cellulare e la sua mutazione influenza non solo le cellule neurali, ma anche le cellule non neurali del cervello, comprese le cellule endoteliali associate al sistema vascolare.
L’integrità vascolare è fondamentale per il mantenimento dell’omeostasi cerebrale e la sua alterazione può essere collegata alla patologia delle malattie neurodegenerative, ma non è stato dimostrato un effetto non neurogeno, come la relazione tra alterazione vascolare e patogenesi della RTT. “In questo studio, abbiamo sviluppato un modello di rete microvascolare utilizzando cellule staminali pluripotenti indotte (iPS) derivate da pazienti affetti da RTT, portatrici della mutazione MeCP2[R306C] o MeCP2[R168X], per studiarne l’impatto vascolare precoce sullo sviluppo“.
Ricercatori del MIT hanno scoperto che due comuni mutazioni genetiche che causano la sindrome di Rett innescano ciascuna una catena molecolare di eventi che compromette l’integrità strutturale dei vasi sanguigni cerebrali in via di sviluppo, rendendoli permeabili. Lo studio riconduce il problema alla sovraespressione di un particolare microRNA (miRNA-126-3p) e dimostra che ridurre i livelli di miRNA aiuta a riparare il difetto vascolare.
La sindrome di Rett è un grave disturbo dello sviluppo che colpisce sia il cervello che il corpo. È causata da diverse mutazioni nel gene MECP2, ampiamente espresso, ma i primi sintomi non si manifestano fino a quando i bambini affetti (per lo più femmine) non raggiungono i 2-3 anni di età. Poiché questo è un momento critico nello sviluppo dei vasi sanguigni del cervello, i neuroscienziati del Picower Institute for Learning and Memory del MIT hanno avviato uno studio per modellae come due mutazioni comuni ma distinte di MeCP2 possano influenzare lo sviluppo vascrolare e contribuire alla profonda patologia neurologica della malattia.
Per condurre la ricerca pubblicata su Molecular Psychiatry, l’autore principale, il ricercatore Tatsuya Osaki e l’autore senior, il Professor Mriganka Sur, hanno sviluppato colture avanzate di tessuti umani per modellare lo sviluppo dei vasi, con e senza mutazioni MeCP2. Le colture non solo hanno permesso loro di modellare e osservare da vicino come le mutazioni influenzassero i vasi, ma hanno anche permesso loro di analizzare a livello molecolare i problemi osservati e quindi di testare un intervento efficace.
“È stato dimostrato il ruolo dei microRNA nella sindrome di Rett, ma ora dimostrare che il miRNA-126-3p è in realtà a valle di MeCP2 e direttamente implicato nella disfunzione delle cellule endoteliali è un pezzo importante del puzzle della sindrome di Rett”, ha affermato Sur, Professore di neuroscienze presso il Picower Institute e il Dipartimento di scienze cognitive e del cervello del MIT.
Costruzione di recipienti e individuazione di perdite
Grazie ad anni di esperienza nell’ingegneria tissutale, tra cui il periodo trascorso come postdoc nel laboratorio del coautore e Professore di ingegneria meccanica e ingegneria biologica del MIT Roger D. Kamm, Osaki ha costruito “reti microvascolari tridimensionali” utilizzando cellule staminali pluripotenti indotte umane (cellule iPS) donate da pazienti affetti dalla sindrome di Rett.
Le cellule donate sono state indotte a trasformarsi in cellule staminali e poi in cellule endoteliali (la spina dorsale dei vasi sanguigni). Inserite in un gel e mescolate con cellule fibroblastiche, le cellule endoteliali si sono autoassemblate in reti di tubi, che Osaki ha poi collegato alla microfluidica per garantire la circolazione.
Un gruppo di colture presentava la mutazione R306C. Osaki ha creato un microcircolo di controllo geneticamente identico, ma privo della mutazione. Un altro gruppo di colture presentava la mutazione R168X. E ancora, Osaki ha accoppiato quest’ultimo con una coltura di controllo identica, fatta eccezione per la mutazione, utilizzando la tecnica CRISPR.
“Il team di ricerca ha scelto queste due mutazioni perché sono entrambe relativamente comuni, ma influenzano il gene MeCP2 in modo diverso”, ha affermato Sur. La scoperta che ciascuna di queste distinte mutazioni che causano la sindrome di Rett ha portato alla sovraregolazione del miRNA-126-3p e alla compromissione dell’integrità dei vasi sanguigni suggerisce che i problemi vascolari siano effettivamente una caratteristica centrale della malattia.
“C’è qualcosa in comune tra queste mutazioni“, ha affermato Sur.
In particolare, i test di laboratorio hanno dimostrato che i vasi che ospitavano entrambe le mutazioni presentavano una ridotta espressione di una proteina chiamata ZO-1, fondamentale per garantire che le giunzioni tra le cellule endoteliali nei vasi sanguigni formino una tenuta stagna (come la malta di un pavimento piastrellato). ZO-1 non si localizzava nemmeno in quelle giunzioni. In effetti, ulteriori test hanno dimostrato che le colture di vasi con mutazione Rett erano relativamente permeabili rispetto ai controlli.
Carenze simili sono state evidenziate in un’altra coltura cellulare creata dal team, in cui sono stati aggiunti astrociti per simulare ancora più fedelmente la barriera emato-encefalica (BBB), che regola rigorosamente ciò che entra o esce dai vasi sanguigni e dal cervello. Si sospetta ampiamente che i problemi alla BBB contribuiscano a malattie neurodegenerative come l’Alzheimer, la malattia di Huntington, la SLA e la demenza frontotemporale.
Per comprendere meglio come i problemi vascolari possano compromettere la funzione neurale nella sindrome di Rett, i ricercatori hanno esposto i neuroni al terreno di coltura delle loro colture vascolari di Rett. Queste cellule nervose hanno mostrato una ridotta attività elettrica, un possibile segno che le secrezioni delle cellule endoteliali di Rett stavano compromettendo la funzionalità dei neuroni.
Catturare un colpevole
In generale, il ruolo di MeCP2 è quello di reprimere l’espressione di altri geni. Gli scienziati si aspettavano, quindi, che quando MeCP2 fosse compromesso da mutazioni, il risultato sarebbe stata la sovraespressione di molti geni. Eppure, ZO-1 risultava sottoregolato. “Qualcosa doveva pur essere responsabile di questo e i miRNA erano un sospettato“, ha detto Osaki, “perché funzionano come regolatori dell’espressione genica”.
“Ecco perché abbiamo ipotizzato che dovesse esserci un mediatore tra la mutazione MeCP2, la downregulation di ZO-1 e l’aumento della permeabilità della barriera emato-encefalica“, ha detto Osaki. “Ci siamo concentrati sui microRNA”.
Infatti, profilando i miRNA nelle colture di Rett e nei controlli, gli scienziati hanno scoperto che il miRNA-126-3p era sovraespresso. E sequenziando l’RNA, il team ha identificato ulteriori vie molecolari necessarie a supportare l’integrità vascolare che risultavano disregolate nelle colture di Rett.
Leggi anche:Sindrome di Rett: riattivazione dei geni silenti offre speranza
Mentre il sequenziamento e il profilo associavano l’aumento della regolazione del miRNA-126-3p alla catena molecolare alterata degli eventi, Osaki e Sur cercavano prove più definitive. Per ottenerle, hanno trattato le colture con mutazione Rett con un “antisenso”, una molecola che riduce i livelli di miRNA-126-3p. Ciò ha portato a un aumento dell’espressione di ZO-1 e a un parziale ripristino della funzione di barriera delle cellule endoteliali, ovvero una minore permeabilità, nelle colture vascolari. L‘abbattimento dell’espressione del miRNA ha anche ripristinato i percorsi molecolari che gli scienziati stavano monitorando verso stati più sani.
A quanto pare esiste un farmaco che inibisce il miR-126, chiamato miRisten, attualmente in fase di sperimentazione clinica per la leucemia. Osaki e Sur affermano di volerlo somministrare a topi che simulano la sindrome di Rett per vedere se funziona.
Fonte: Molecular Psychiatry