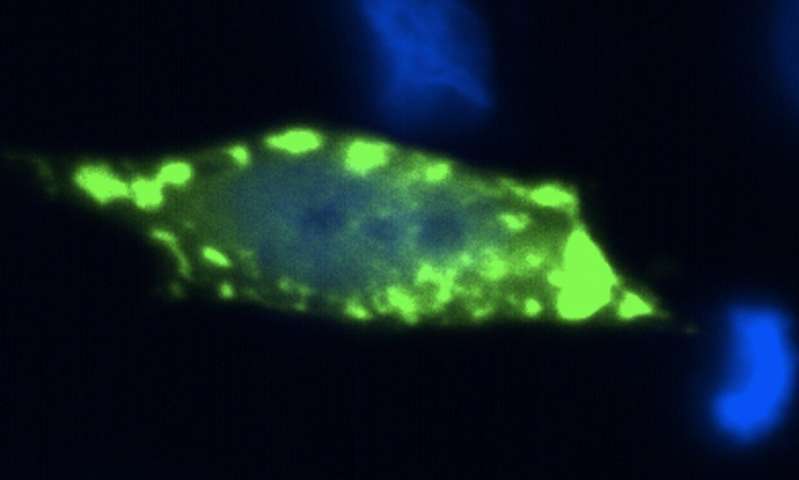
Immagine, cellule umane che producono aggregati della proteina Sod1 (in verde). Credito: Aline Brasil, Elis Eleutherio e Tiago Outeiro.
Un team guidato dal ricercatore brasiliano Elis Eleutherio, Professore presso l’Università Federale di Rio de Janeiro, in collaborazione con Tiago Outeiro, presso l’Università di Gottinga, in Germania, ha compiuto importanti progressi nella comprensione della conformazione e dell’accumulo di alcune proteine coinvolte nella sclerosi laterale amiotrofica (SLA).
“Riteniamo che l’accumulo di proteine sia un importante segno distintivo della SLA e non capiamo ancora perché le proteine si comportano male e formano aggregati durante la malattia“, spiega il Prof. Eleutherio.
La SLA è una malattia neurodegenerativa progressiva e devastante che colpisce da 1 a 3 individui su 100.000 ed è più diffusa nelle persone di età compresa tra 55 e 75 anni. La malattia colpisce principalmente una popolazione di neuroni nota come “motoneurone”. I pazienti soffrono di paralisi motoria irreversibile e diventano incapaci di parlare, deglutire o respirare mentre la malattia progredisce.
La maggior parte dei casi di SLA sono sporadici, senza origine genetica definita e solo la minoranza è familiare, con alterazioni genetiche associate note. Alcune forme familiari di SLA (fSLA) sono associate ad alterazioni genetiche nel gene che codifica per una proteina nota come superossido dismutasi 1 (Sod1), che provoca alterazioni nel ripiegamento e nella funzione della proteina.
Vedi anche, Farmaco approvato dalla FDA mostra risultati promettenti contro la SLA.
Lo studio, pubblicato sulla rivista Proceedings of National Academy of Sciences (PNAS), ha permesso agli scienziati di comprendere l’interazione tra la proteina normale e quella mutante, che provoca l’alterazione dell’accumulo di proteine nella cellula, ma compromette anche la funzione di Sod1 contribuendo così allo sviluppo della malattia. Per il gruppo, questa scoperta apre nuove prospettive per il trattamento della SLA.
Sod1 è una proteina che svolge un ruolo, tra gli altri, nella protezione contro il danno ossidativo nelle nostre cellule. In alcuni casi di SLA, la proteina Sod1 alterata si accumula all’interno delle cellule neuronali e, secondo i ricercatori, provoca danni ai neuroni, portando alla loro morte. È importante sottolineare che le normali proteine Sod1 presenti in casi sporadici di SLA possono anche ripiegarsi e accumularsi erroneamente, suggerendo che questo è un problema centrale nella SLA.
Nello studio, i ricercatori hanno utilizzato semplici modelli sperimentali, come il lievito utilizzato per produrre birra, vino e pane e cellule umane, al fine di comprendere meglio il contesto dell’aggregazione proteica nella malattia. Hanno anche usato una strategia che imita il contesto genetico delle fSLA o SLA familiare in cui la maggior parte dei pazienti trasporta una copia della normale proteina Sod1 e una copia con un’alterazione genetica. “Nei pazienti, riteniamo che la presenza di una copia mutante di Sod1 modifichi il comportamento della copia normale”, spiega la Dott.ssa Aline Brasil, la prima autrice dello studio.
“Sfruttando i nuovi strumenti di manipolazione genetica e i potenti approcci di imaging molecolare che consentono la visualizzazione diretta dei complessi proteici nella cellula (una tecnica nota come BiFC), siamo stati in grado di rilevare” etero-complessi “formati da normali e anormali ( mutanti) proteine Sod1 “, ha affermato il Prof. Outeiro, leader del team tedesco che ha partecipato allo studio.
La ricerca apre nuove prospettive di intervento terapeutico che gli autori sperano di continuare a esplorare nel prossimo futuro, come la rimozione specifica della proteina mutante Sod1.
Fonte, PNAS