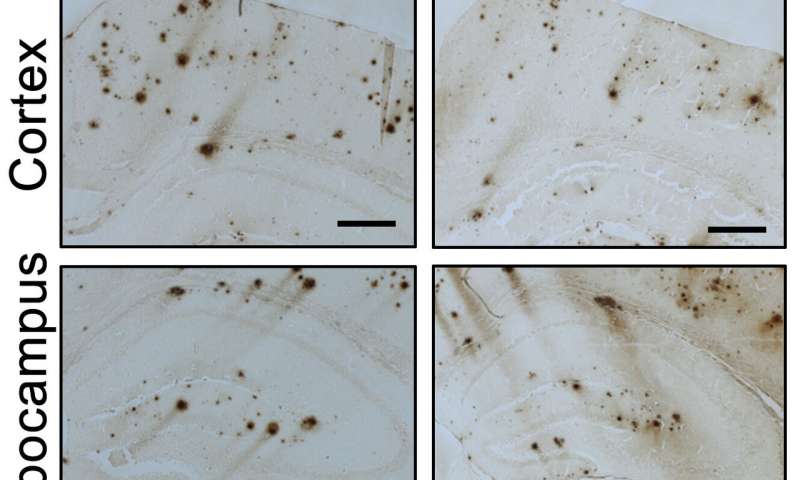
Immagine, i topi trattati con Idazoxan, che blocca un recettore della noradrenalina, si sono comportati in modo simile ai topi di controllo nonostante abbiano ancora placche beta-amiloide nel cervello. Credito: F. Zhang et al., Science Translational Medicine (2019)
In esperimenti preclinici, i ricercatori dell’Università di Alabama a Birmingham hanno rivelato un pezzo mancante chiave del puzzle del morbo di Alzheimer. Ciò ha consentito esperimenti con un farmaco esistente che ha ridotto drasticamente la patologia e i sintomi dell’Alzheimer in due modelli murini, offrendo potenzialmente un trattamento immediato per questa malattia devastante.
I ricercatori hanno scoperto che l’Alzheimer è attivato da una segnalazione di noradrenalina difettosa.
La ricerca è stata pubblicata oggi sulla rivista Science Translational Medicine. Comprende l’analisi del tessuto cerebrale umano e dati clinici longitudinali a supporto dei dati del modello murino in vivo. “Il nostro studio fornisce approfondimenti traslazionali sui meccanismi alla base della tossicità delle proteine beta-amiloide, che possono avere forti implicazioni per la futura progettazione di farmaci”, ha affermato Qin Wang, M.D., Ph.D. “Abbiamo identificato un’interazione recettoriale accoppiata tra proteina amiloide / beta / proteine G ( proteine G che rilevano molecole all’esterno di una cellula ) che rappresenta un bersaglio terapeutico attraente e specifico per la malattia di Alzheimer”. È interessante notare che il meccanismo patologico trovato può anche spiegare il fallimento di numerosi studi clinici sull’Alzheimer che miravano alla riduzione del colpevole del morbo di Alzheimer, l’accumulo di proteine amiloidi nel cervello.
Alla UAB School of Medicine, Wang è Professore presso il Dipartimento di Biologia cellulare, dello sviluppo e dell’integrazione. “È ampiamente riconosciuto”, afferma Wang, “che l‘accumulo di oligomeri beta-amiloidi nel cervello agisce come un fattore scatenante per indurre cambiamenti patologici nella proteina tau e che l’alterata proteina tau è il proiettile che prende di mira e uccide i neuroni nella malattia di Alzheimer. Tuttavia, il percorso che collega questi due era sconosciuto”.
Wang e colleghi hanno scoperto che gli oligomeri beta-amiloidi dirottano la segnalazione della noradrenalina ai neuroni cerebrali che reindirizza falsamente questo segnale per attivare una chinasi chiamata GSK3-beta. Quell’enzima chinasi attivato, a sua volta, produce proteine tau iper-fosforilate tossiche per i neuroni.
Questo ricablaggio della segnalazione della noradrenalina avviene in corrispondenza di un recettore della membrana cellulare sulla superficie dei neuroni chiamato recettore adrenergico alfa-2A. Questo recettore fa parte di una vasta famiglia di recettori accoppiati a proteine G che rilevano molecole all’esterno di una cellula e quindi attivano un segnale interno che provoca una risposta cellulare.
Wang e colleghi hanno scoperto che mentre una certa concentrazione di oligomeri beta-amiloidi può attivare la chinasi GSK3-beta, la presenza di noradrenalina ha sensibilizzato tale attivazione fino a due ordini di grandezza.
Pertanto, i ricercatori UAB ipotizzano che le concentrazioni nanomolari di oligomeri beta-amiloidi nel cervello umano inducano una cascata patogena GSK3-beta / tau nelle prime fasi della malattia di Alzheimer. Questa teoria suggerisce perché diversi studi clinici per ridurre i livelli di oligomeri beta-amiloidi nei pazienti con malattia di Alzheimer hanno fallito: non possono ridurre i livelli di amiloide a concentrazioni così basse.
Dettagli dello studio
Il recettore adrenergico alfa-2A normalmente funziona in questo modo: ha un sito di legame per la neurotrasmettitrice noradrenalina e quel legame attiva un processo di segnalazione che mobilita il cervello e il corpo per l’azione. I ricercatori UAB hanno scoperto che gli oligomeri beta-amiloidi si legano a un sito separato sul recettore adrenergico alfa-2A, distinto dal sito per il legame con noradrenalina. Questo avvia il dirottamento patologico. Tale legame in un secondo sito è chiamato legame allosterico. Nei recettori accoppiati a proteine G, i ligandi allosterici sono noti per alterare spesso la segnalazione del recettore come parte della normale fisiologia. Dopo che i ricercatori hanno realizzato il legame allosterico, hanno cercato di vedere quale chinasi potesse essere attivata da quel legame, ed è così che hanno identificato GSK3-beta.
Alcuni dati clinici supportano questo meccanismo.
I ricercatori hanno scoperto nelle cortecce prefrontali post mortem dei pazienti con malattia di Alzheimer c’era stato un aumento significativo dell’attività del recettore adrenergico alfa-2A, rispetto ai controlli a bassa patologia. Inoltre, l’analisi epidemiologica dei casi dal Centro di coordinamento nazionale dell’Alzheimer ha mostrato che l’assunzione del farmaco Clonidina – un attivatore del recettore adrenergico alfa-2A utilizzato per abbassare la pressione sanguigna – ha peggiorato la funzione cognitiva nei pazienti con deficit cognitivi. Inoltre, gli effetti avversi della Clonidina sono stati più forti nei pazienti con demenza più grave. L’uso della clonidina non ha avuto effetti su soggetti con cognizione normale.
Wang e colleghi hanno testato un farmaco esistente – Idazoxan – in un modello murino di Alzheimer. Idazoxan è un antagonista del recettore adrenergico alfa-2A ed è stato oggetto di studi clinici per la depressione. L’ipotesi era che il blocco di Idazoxano del recettore adrenergico alfa-2A in presenza di patologia beta-amiloide avrebbe mostrato potenziale terapeutico. Ciò è stato confermato nei topi modello di Alzheimer.
I topi sono stati trattati con Idazoxan per otto settimane a partire dall’età di 8 mesi, quando le placche beta-amiloide sono già presenti nel cervello e il recettore adrenergico alfa-2A mostra una maggiore attività rispetto ai controlli.
I ricercatori UAB hanno scoperto che:
1) Idazoxan ha invertito l’iperattivazione di GSK3-beta nel cervello del topo, fornendo un supporto aggiuntivo per il ruolo critico del recettore adrenergico alfa-2A nella mediazione dell’attivazione indotta da beta-amiloide-GSK in vivo;
2) Nella corteccia cerebrale dei topi modello Alzheimer trattati con Idazoxano, l’estensione del carico di beta-amiloide era inferiore, dimostrando che il blocco del recettore adrenergico alfa-2A rallentava la progressione della patologia beta-amiloide;
3) Il trattamento con Idazoxan ha ridotto la densità delle cellule infiammatorie della microglia, suggerendo una riduzione della neuroinfiammazione;
4) Il trattamento con Idazoxan ha ridotto l’iperfosforilazione di tau, suggerendo che il blocco del recettore adrenergico alfa-2A allevia efficacemente la patologia tau indotta da beta-amiloide;
5) I topi modello Alzheimer trattati con Idazoxan hanno eseguito compiti quasi come i topi normali e significativamente in modo migliore rispetto ai topi modello Alzheimer non trattati, in due test per la funzione cognitiva.
“Questi dati dimostrano collettivamente che bloccare la segnalazione della noradrenalina attraverso il recettore adrenergico alfa-2A è una strategia efficace per migliorare i deficit patologici e cognitivi associati alla beta-amiloide“, ha detto Wang. “I bloccanti del recettore adrenergico alfa-2A come Idazoxan sono stati sviluppati per l’uso in altri disturbi e il riutilizzo di questi farmaci potrebbe essere una strategia potenzialmente efficace e facilmente disponibile per il trattamento della malattia di Alzheimer”, ha aggiunto Wang. “Inoltre, i nostri dati suggeriscono che l’interazione del recettore adrenergico amiloide-beta / alfa-2A è un bersaglio terapeutico attraente e specifico per la malattia di Alzheimer perché il recettore alfa-2A adrenergico / GSK3-beta / tau può essere attivato solo alla presenza di oligomeri beta-amiloidi”.
“Il target diretto dell’interfaccia del recettore adrenergico amiloide beta / alfa-2A allosterico non interferirebbe con le normali funzioni del recettore adrenergico alfa-2A”, ha detto Wang, “e quindi avrebbe meno probabilità di provocare complicazioni associate ad un lungo periodo di dosaggio necessario per il trattamento della malattia di Alzheimer “.