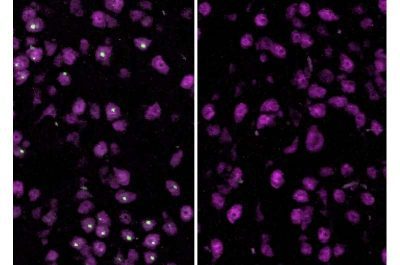
Malattia di Huntington-immagine: le cellule cerebrali di topo non trattate (a sinistra), portatrici della mutazione della malattia di Huntington, mostrano grandi aggregati proteici (in verde). Nelle cellule (a destra) trattate con oligonucleotidi antisenso che prendono di mira l’huntingtina 1a, gli aggregati sono quasi scomparsi. Crediti: Laboratorio di Jeffrey Carroll / UW Medicine Neurology
Un nuovo studio condotto sui topi suggerisce che i trattamenti mirati a un frammento della proteina mutante responsabile della malattia di Huntington potrebbero essere più efficaci rispetto a quelli – attualmente in fase di sperimentazione clinica – che agiscono sull’intera proteina lasciando intatto questo frammento.
I risultati sono stati pubblicati sulla rivista Science Translational Medicine.
“Le basi scientifiche dei nostri risultati sono solide“, ha affermato l’autore senior Jeffrey Carroll, Professore associato di neurologia presso la Facoltà di Medicina dell’Università di Washington a Seattle. “Per avere successo, potremmo dover sviluppare nuove terapie che prendano di mira anche questa specifica regione della proteina“.
La malattia di Huntington è causata da una mutazione in un gene chiamato huntingtina. La mutazione produce una proteina anomala che si accumula nelle cellule cerebrali. Qui interferisce con un’ampia gamma di funzioni cellulari e forma grandi aggregati della proteina che portano alla morte cellulare. Una persona deve ereditare una sola copia del gene mutato da uno dei genitori per contrarre la malattia.
Le persone affette dalla malattia di Huntington in genere iniziano a manifestare i primi sintomi intorno ai 40 anni, sebbene l’esordio possa essere precoce o tardivo. I sintomi iniziali includono movimenti incontrollati (chiamati corea di Huntington), goffaggine e problemi di equilibrio. Con il progredire della malattia, chi ne è affetto perde la capacità di camminare, parlare e deglutire e alla fine necessita di assistenza 24 ore su 24. Cambiamenti di personalità e demenza sono comuni nelle fasi avanzate della malattia. La condizione progredisce inesorabilmente ed è fatale entro 10-15 anni dalla comparsa dei sintomi. Circa 41.000 americani sono affetti dalla malattia di Huntington e oltre 200.000 sono a rischio di svilupparla.
Spiegano gli autori:
“La malattia di Huntington (HD) è una malattia neurodegenerativa autosomica dominante causata dall’espansione di una ripetizione CAG codificante per la glutammina nel gene dell’huntingtina ( HTT ) oltre una soglia critica. L’esordio della HD si verifica generalmente nella mezza età, con una serie progressiva di sintomi cognitivi, affettivi e motori, con l’età di esordio strettamente legata alla dimensione della ripetizione CAG. Le evidenze genetiche suggeriscono che la HD derivi da un guadagno tossico di funzione, sebbene non sia ancora chiaro quale/i perturbazione/i della funzione dell’HTT determini la tossicità o se il guadagno tossico di funzione sia interamente neomorfico. Questi squilibri nell’omeostasi cellulare sono particolarmente tossici per i neuroni, con una marcata perdita di neuroni spinosi medi (MSN) nello striato, sebbene l’origine di questa specificità cellulare non sia completamente compresa. Date queste lacune nelle nostre conoscenze, rimane una sfida distinguere quali cambiamenti molecolari e fisiologici nei neuroni affetti da HD siano parte di una cascata patogenetica causale e quali siano epifenomeni”.
Attualmente non esiste una cura efficace, ma sono in fase di sperimentazione diverse terapie sperimentali. L’approccio più promettente consiste nel prevenire la produzione della proteina anomala sabotando il processo mediante il quale le istruzioni del DNA codificate nel gene vengono tradotte in proteina.
In questo processo, le istruzioni del DNA del gene vengono prima copiate in una forma di RNA, chiamata RNA messaggero (mRNA), che la cellula legge per assemblare la proteina. I ricercatori possono interrompere questo processo introducendo una breve sequenza di DNA chiamata oligonucleotide antisenso che si lega a un sito specifico sul filamento di mRNA. Ciò induce gli enzimi della cellula a tagliare il filamento in quel punto, impedendo così alla cellula di produrre la proteina completa. L’mRNA tagliato e la proteina incompleta vengono quindi eliminati dalla cellula.
Nel loro nuovo studio, Carroll e i suoi colleghi inizialmente volevano confrontare i trattamenti con oligonucleotidi antisenso che riducevano la produzione di tutte le proteine huntingtina, sia quelle normali che quelle mutanti, con un trattamento che bloccava la produzione solo della versione mutante.
Come si è scoperto, il trattamento più efficace sul tipo di topo studiato si legava all’mRNA molto vicino all’inizio del filamento. Ciò significava che non solo sopprimeva la produzione dell’intera proteina, ma anche quella di un brevissimo segmento della proteina chiamato huntingtina 1a. L’huntingtina 1a è nota per essere tossica per le cellule nervose, ma il suo ruolo nella malattia di Huntington non è ancora del tutto chiaro.
Dopo aver trattato dei topi portatori di una copia del gene aberrante, i ricercatori hanno valutato l’efficacia dei trattamenti analizzando l’espressione di oltre 150 geni coinvolti nella malattia di Huntington. Hanno inoltre verificato la presenza di aggregati proteici, un segno distintivo della malattia.
Il trattamento che bloccava la produzione dell’intera proteina, ma non dell’huntingtina 1a, ha avuto scarso effetto. Tuttavia, il trattamento con l‘oligonucleotide antisenso che impediva la produzione dell’huntingtina 1a si è rivelato straordinariamente efficace. Ad esempio, l’espressione di circa il 55% dei geni normalmente coinvolti nella malattia di Huntington è tornata ai livelli basali e la formazione di aggregati è stata quasi completamente eliminata.
Leggi anche: Malattia di Huntington curata per la prima volta con la terapia genica
“Quando ho osservato al microscopio le cellule dei topi trattati, ho pensato di aver commesso un errore perché inizialmente non riuscivo a trovare alcun aggregato proteico”, ha affermato Robert Bragg, primo autore dello studio e ricercatore presso il laboratorio di Carroll. “Ho dovuto cercare con molta attenzione per trovarne anche solo uno o due“. “Sembra che se si riduce l’espressione della proteina di Huntington completa, ma l’huntingtina 1a continua a essere espressa, non si ottenga alcun risultato“, ha affermato Bragg. “A quanto pare, è necessario ridurre drasticamente l’espressione dell’huntingtina 1a per ottenere un effetto significativo“.
Fonte: Science Translational Medicine