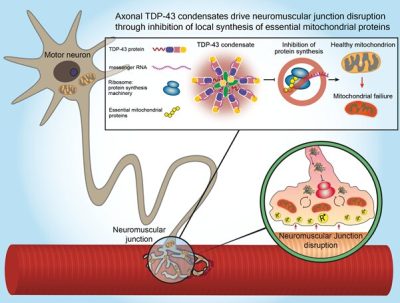
(SLA-Immagine: illustrazione che mostra la proteina TDP-43 che si accumula in modo distruttivo nelle estensioni dei nervi motori, in particolare nelle giunzioni neuromuscolari dei pazienti con SLA, dove intrappola le molecole di RNA messaggero e impedisce la sintesi di proteine essenziali per la funzione mitocondriale. Credito: Università di Tel Aviv).
Un gruppo di ricerca della Facoltà di Medicina Sackler e della Scuola di Neuroscienze Sagol dell’Università di Tel Aviv ha scoperto, per la prima volta, il meccanismo biologico che causa la distruzione dei nervi nella malattia neurodegenerativa SLA. Lo studio innovativo, condotto dal Prof. Eran Perlson e dai dottorandi Topaz Altman e Ariel Ionescu, suggerisce che il decorso di questa malattia mortale può essere ritardato e persino invertito nelle sue fasi iniziali. Lo studio è stato condotto in collaborazione con il Dr. Amir Dori, Direttore della clinica per le malattie neuromuscolari presso lo Sheba Medical Center.
I risultati dello studio sono stati pubblicati su Nature Communications.
La SLA è il tipo più comune di malattia del motoneurone. Causa paralisi e atrofia muscolare. Una persona su 400 avrà la malattia, eppure non esiste una cura efficace. I pazienti con SLA perdono gradualmente la capacità di controllare i movimenti muscolari volontari, sviluppano la paralisi completa e alla fine perdono la capacità di respirare autonomamente. L’aspettativa di vita media dei pazienti con SLA è attualmente solo di circa tre anni.
“A tutt’oggi non è chiaro cosa causi la malattia”, spiega il Prof. Perlson. “Solo il 10% circa dei pazienti ha un background familiare con mutazioni genetiche note, ma il restante 90% è un mistero. La paralisi causata dalla malattia deriva da un danno ai motoneuroni, che porta alla degenerazione delle terminazioni nervose e alla perdita di innervazione muscolare. Ciò porta di conseguenza alla degenerazione dei nervi e alla morte dei motoneuroni nel midollo spinale, tuttavia fino ad ora non siamo riusciti a comprendere il meccanismo biologico di base che causa il danno iniziale dietro questa cascata viziosa”.

Per risolvere il mistero, i ricercatori dell’Università di Tel Aviv si sono concentrati su una proteina chiamata TDP-43, che in studi precedenti aveva dimostrato di accumularsi in quantità e localizzazione insolite nel cervello di circa il 95% di tutti i pazienti con SLA. Il Prof. Perlson e il suo team hanno rivelato un nuovo legame biologico tra l’accumulo della proteina e la degenerazione delle sinapsi tra le terminazioni dei motoneuroni e i muscoli, chiamate giunzioni neuromuscolari, che traducono i comandi neurali in movimenti fisici. Nelle biopsie muscolari prelevate da pazienti con SLA i ricercatori hanno scoperto che la proteina tossica si accumula anche in prossimità di queste giunzioni neuromuscolari durante le prime fasi della malattia e prima che i pazienti sviluppino sintomi gravi. In una serie di esperimenti eseguiti dai ricercatori, sia in cellule di pazienti affetti da SLA che in animali modello geneticamente modificati, i ricercatori hanno scoperto che l’accumulo della proteina TDP-43 nella giunzione neuromuscolare inibisce la capacità di sintetizzare localmente proteine essenziali per l’attività mitocondriale,che fornisce il potere di processi cellulari fondamentali. La disfunzione dei mitocondri nelle terminazioni nervose porta alla rottura della giunzione neuromuscolare e infine alla morte dei motoneuroni. “È importante prima capire la complessità spaziale dei motoneuroni”, afferma il Prof. Perlson. La disfunzione dei mitocondri nelle terminazioni nervose porta alla rottura della giunzione neuromuscolare e infine alla morte dei motoneuroni.
Vedi anche:SLA: prime immagini dettagliate della molecola responsabile
“Abbiamo mostrato che nella SLA questa proteina esce dal nucleo e si accumula in tutta la cellula e in particolare nella giunzione neuromuscolare. Poiché la funzione dei motoneuroni dipende da queste giunzioni neuromuscolari, ci siamo resi conto che questo risultato poteva essere di importanza critica. Abbiamo scoperto che gli accumuli formati dalla proteina TDP-43 nelle giunzioni neuromuscolari intrappolano le molecole di RNA e impediscono la sintesi di proteine essenziali alla funzione mitocondriale. I mitocondri sono organelli presenti nelle cellule e sono i principali fornitori di energia per numerosi processi cellulari, inclusa la trasmissione neurale. La condensazione della proteina TDP-43 nelle giunzioni neuromuscolari provoca un grave depauperamento energetico, previene la riparazione mitocondriale nei motoneuroni nel midollo spinale”.
Per confermare le loro scoperte, i ricercatori dell’Università di Tel Aviv hanno deciso di utilizzare una molecola sperimentale recentemente pubblicata da un gruppo di ricercatori statunitensi per essere sviluppata per un altro scopo: migliorare la rigenerazione neurale dopo il danno causato dallo smontaggio dei condensati proteici nelle estensioni neurali. I ricercatori hanno dimostrato che questa molecola potrebbe anche disassemblare i condensati della proteina assonale TDP-43 nelle cellule dei pazienti con SLA e che questo processo ha migliorato la capacità di produrre proteine essenziali, potenziato l’attività mitocondriale e prevenuto la degenerazione della giunzione neuromuscolare. Inoltre, negli animali modello, i ricercatori hanno dimostrato che invertendo l’accumulo di TDP-43 nei nervi e nella giunzione neuromuscolare è stato possibile recuperare le giunzioni neuromuscolari degenerate e riabilitare quasi completamente gli animali modello malati.

“Nel momento in cui abbiamo indotto lo smontaggio dei condensati proteici TDP-43, è stata recuperata la capacità dei nervi di produrre proteine, in particolare la sintesi di proteine essenziali per l’attività mitocondriale.Tutto ciò ha permesso ai nervi di rigenerarsi“, riassume il Prof. Perlson. “Siamo stati in grado di dimostrare, attraverso mezzi farmacologici e genetici, che i nervi motori possono rigenerarsi e che i pazienti possono avere speranza. Infatti, abbiamo individuato il meccanismo di base, così come le proteine responsabili della rottura dei nervi da muscoli e della loro degenerazione. Questa scoperta può portare allo sviluppo di nuove terapie che potrebbero dissolvere la proteina TDP-43 o aumentare la produzione di proteine essenziali alla funzione mitocondriale, e quindi guarire le cellule nervose prima del danno irreversibile che si verifica nel midollo spinale. “Stiamo affrontando il problema dall’altra parte, nella giunzione neuromuscolare. E se in futuro si potesse diagnosticare e intervenire con sufficiente anticipo, forse sarà possibile inibire la degenerazione distruttiva nei muscoli dei malati di Sla”, dice il ricercatore Perlson.
Lo studio è una collaborazione internazionale con eminenti scienziati provenienti da Germania, Francia, Inghilterra e Stati Uniti, con l’assistenza di Tal Gardus Perry e Amjad Ibraheem del laboratorio del Prof. Perlson.
Fonte: Nature