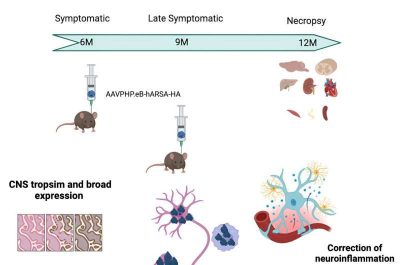
Leucodistrofia metacromatica-Immagine:graphical abstract. Credit: Molecular Therapy – Methods & Clinical Development.
La leucodistrofia metacromatica è una malattia genetica rara che colpisce principalmente i bambini piccoli e provoca gravi sintomi neurologici accompagnati da una perdita delle capacità motorie e intellettuali. Al Paris Brain Institute, Françoise Piguet e i suoi colleghi, hanno sviluppato un trattamento di terapia genica in grado di correggere l’anomalia primaria osservata nella malattia: l’accumulo di solfatidi nel cervello e nel midollo spinale. Efficace nei topi, come dimostrato dai risultati pubblicati su Molecular Therapy—Methods & Clinical Development, questa tecnica apre la strada agli studi clinici.
La leucodistrofia metacromatica colpisce da una a nove persone su 100.000 e si manifesta principalmente durante l’infanzia e l’adolescenza, con il 10-20% dei casi osservati in età adulta. La malattia è ereditaria, ma richiede che entrambi i genitori siano portatori del gene ARSA mutato, che controlla la produzione dell’enzima arilsolfatasi A.
L’esaurimento di questo enzima nel corpo porta ad un accumulo anormale di lipidi specifici (solfatidi) nella sostanza bianca del sistema nervoso centrale, dei nervi periferici, dei reni e della cistifellea.
La conseguenza? Una perdita della guaina mielinica, che garantisce la corretta conduzione dei segnali nervosi nel cervello e nel midollo spinale, e una risposta infiammatoria acuta che danneggia le cellule nervose. Di conseguenza, i pazienti manifestano sintomi gravemente debilitanti come disturbi motori, visivi e uditivi, un deterioramento delle capacità intellettuali e difficoltà ad esprimersi.
La leucodistrofia metacromatica progredisce particolarmente rapidamente nei bambini e porta a morte prematura, da qui l’urgente necessità di sviluppare trattamenti efficaci.
“Per questi giovani pazienti i cui sintomi si sono già manifestati, la terapia genica, che prevede l’iniezione di un gene sano nell’organismo utilizzando un virus innocuo, è un modo molto promettente per ritardare o arrestare la progressione della malattia“, afferma Françoise Piguet, ricercatrice e capo dell’unità di innovazione e sviluppo tecnologico Genov presso il Paris Brain Institute.
“Questa tecnica rende possibile fare in modo che le cellule nervose esprimano la proteina mancante, in questo caso l’enzima arilsolfatasi A. Tuttavia, affinché la terapia genica in vivo sia efficace, è necessario superare un vincolo cruciale: consentire al carico di materiale genetico di attraversare la barriera ematoencefalica, che impedisce alle sostanze indesiderate che circolano nel sangue di raggiungere il cervello”.
Affrontare la barriera emato-encefalica
Per il loro studio, Françoise Piguet e la sua équipe hanno scelto un tipo specifico di virus “adeno-associato” (AAVPHP.eB) le cui proprietà gli permettono di attraversare facilmente questa barriera e sono innocue per l’organismo. I ricercatori lo hanno utilizzato come vettore – o come veicolo – per trasportare una copia del gene funzionale ARSA nel cervello dei topi in cui questo gene era carente.
“Abbiamo somministrato il gene nel farmaco a topi di sei mesi e poi a topi di nove mesi con sintomi più gravi“, aggiunge Françoise Piguet. “Gli effetti del trattamento sono stati poi valutati tre e sei mesi dopo ogni iniezione“.
Spiegano gli autori:
“La leucodistrofia metacromatica (MLD) è una malattia ereditaria da accumulo lisosomiale (LSD) autosomica recessiva causata da un deficit di arilsulfatasi A (ARSA; EC 3.1.6.8). L’enzima lisosomiale ARSA catalizza la degradazione del galattosil-3-solfato ceramide (solfatide), uno dei principali sfingolipidi della mielina.La malattia è caratterizzata dalla degenerazione mielinica sia del sistema nervoso centrale (SNC) che del sistema nervoso periferico (PNS), che clinicamente provoca deficit motori e cognitivi progressivi che portano alla morte prematura. Si distinguono tre forme cliniche, in base all’età di esordio dei sintomi: tardiva infantile (LI; età di esordio prima dei 30 mesi); giovanile, suddiviso in giovanile precoce (EJ; tra 2,5 e 7 anni) e giovanile tardivo (LJ; tra 7 e 16 anni); e forme per adulti (dopo i 16 anni). I livelli di attività ARSA residua sono correlati al tipo e alla gravità dei sintomi. Le forme più frequenti e gravi di MLD sono le forme ad esordio precoce, ovvero LI ed EJ. Questi pazienti sviluppano sintomi neurologici, tra cui disturbi dell’andatura e perdita della parola, intorno ai 2 anni di età e muoiono entro pochi anni dalla comparsa dei sintomi. Attualmente non esiste una terapia approvata per arrestare o ritardare la progressione della malattia una volta che i pazienti sono sintomatici. Tuttavia, tre approcci sono stati studiati negli studi clinici o hanno addirittura ottenuto l’approvazione dell’EMA (Agenzia europea per i medicinali): terapia enzimatica sostitutiva intratecale (IT-ERT), terapia genica ex vivo basata sull’autotrapianto di cellule staminali ematopoietiche modificate con vettore lentivirale (HSC-GT) e terapia genica basata su virus adeno-associati. ERT è una buona opzione, tuttavia, l’ARSA non attraversa in modo efficiente la barriera ematoencefalica (BBB). Sebbene i risultati ottenuti in vitro e nei topi MLD suggeriscano che ARSA sia in grado di attraversare in una certa misura la BEE, l’incrocio inefficiente ostacola la somministrazione adeguata al cervello mediante iniezione endovenosa, pertanto è necessaria la somministrazione intratecale. I risultati di uno studio di fase I-II, condotto su pazienti LI-MLD con sintomi precoci, hanno confermato un buon profilo di sicurezza e, per i pazienti che hanno ricevuto la dose più alta (100 mg ogni 2 settimane), una normalizzazione del contenuto di sulfatide nel fluido cerebrospinale (CSF), nonché una tendenza a un minor declino delle funzioni motorie nel tempo nonostante il degrado“.
I risultati dei ricercatori sono molto incoraggianti. Il gene sano si è diffuso con successo alla popolazione di neuroni bersaglio, che ha iniziato a secernere il prezioso enzima e a correggere l’attività delle cellule vicine, compresi gli oligodendrociti produttori di mielina.
Il risultato?
I livelli di solfatidi sono tornati alla normalità e la neuroinfiammazione è stata significativamente ridotta nel cervello e nel midollo spinale, anche nei topi di nove mesi con malattia più avanzata.
“Questi dati sono sufficientemente solidi da poter ora valutare il trattamento nei primati per organizzare successivamente studi clinici sugli esseri umani“, conclude il ricercatore. “La leucodistrofia metacromatica si manifesta rapidamente nei bambini piccoli e la terapia genica deve fermare quasi immediatamente l’accumulo anomalo di solfatidi. Tutti i nostri sforzi sono dedicati al raggiungimento di questo obiettivo“.