Esiste un gruppo di malattie rare, autosomiche recessive, conosciute come Sindrome da deplezione del DNA mitocondriale (MTDPS) che provoca una significativa riduzione della produzione di DNA mitocondriale (mtDNA) e ATP.
I mitocondri sono responsabili di fornire alla cellula energia sotto forma di ATP attraverso la fosforilazione ossidativa.
Non esiste alcun trattamento per la MTDPS che è tipicamente fatale durante l’infanzia a causa di insufficienza epatica.
I ricercatori della Medical University of South Carolina (MUSC) hanno identificato diversi farmaci candidati per il trattamento della malattia.
I loro risultati, pubblicati online nel numero di Cell Reports del 6 novembre 2018, hanno dimostrato che la nicotinamide adenina dinucleotide (NAD) ha ripristinato in modo significativo la funzione mitocondriale sia in vitro che in modelli animali preclinici.
La Sindrome da deplezione del DNA mitocondriale ha recentemente ricevuto l’attenzione internazionale dopo che un bambino, Charlie Gard, è diventato il protagonista di un caso di “interesse” nel Regno Unito. I medici hanno determinato che Gard aveva una mutazione nella subunità ribonucleoside-difosfatasi reduttasi M2 B (RRM2B), una proteina fondamentale per generare i blocchi costitutivi del mtDNA e una delle varie cause di MTDPS. Sfortunatamente, senza cure o trattamenti disponibili, Gard è morto a luglio 2017, dopo aver combattuto contro la malattia per 11 mesi e 24 giorni.
“Ci siamo resi conto che non esiste un trattamento per queste malattie, quindi abbiamo pensato che potremmo iniziare a utilizzare i nostri sistemi di modelli di cellule staminali per cercare di trovare farmaci o terapie” afferma Stephen A. Duncan, SmartState Chair of Regenerative Medicina al MUSC e autore senior di questo studio.
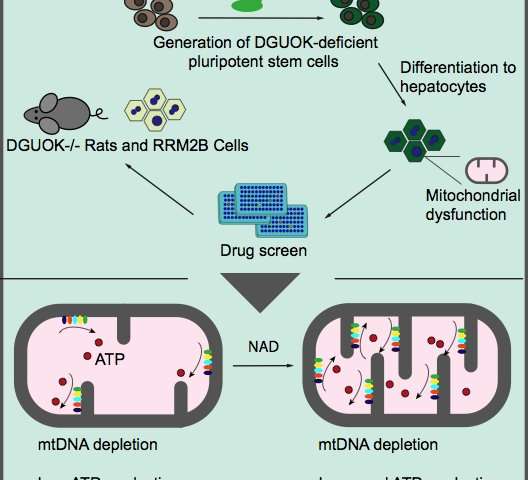
La scoperta di nuovi trattamenti è stata ostacolata dalla mancanza di un modello che imita completamente la patologia della Sindrome da deplezione del DNA mitocondriale. Inoltre, le cellule epatiche primarie dei pazienti sono difficili da ottenere . Per risolvere questo problema, Duncan e il suo team hanno utilizzato CRISPR / Cas9 per esaurire la deossiguanosina chinasi (DGUOK) in cellule staminali pluripotenti indotte dall’uomo. Quindi, usando i protocolli stabiliti nel laboratorio Duncan, i ricercatori hanno differenziato quelle cellule knock-out in cellule simili al fegato.
La MTDPS più comune, che rappresenta il 15-30% dei casi, risulta dalla perdita di DGUOK. Nei pazienti, la perdita di DGUOK riduce i pool di molecole precursori necessarie per sintetizzare mtDNA, con conseguente bassi livelli di mtDNA e diminuiti livelli di ATP. Le cellule con deficienza di DGUOK hanno mostrato una riduzione dei livelli di mtDNA, perdita di produzione di ATP e una morfologia mitocondriale disorganizzata.
Questi fenotipi potrebbero essere invertiti mediante l’aggiunta di precursori per la sintesi di mtDNA e la ri-espressione di DGUOK.
La fase successiva di questo studio ha utilizzato un elenco di farmaci attualmente disponibili sul mercato, per eseguire uno screening fenotipico per farmaci che potrebbero ripristinare i livelli di ATP. Lo schermo ha identificato 15 farmaci, che hanno avuto ripercussioni su una varietà di processi metabolici che hanno aumentato significativamente i livelli di ATP.
Inaspettatamente, il farmaco con il maggiore impatto sulla produzione di ATP è stato la nicotinamide adenina dinucleotide (NAD), un coenzima che colpisce diversi percorsi. I ricercatori hanno continuato a dimostrare che NAD funziona attraverso l’attivazione di una cascata di trascrizione che si traduce in una maggiore espressione delle proteine mitocondriali coinvolte nella produzione di ATP.
“Poiché gli epatociti derivati dalle iPSC ci hanno permesso di realizzare uno schermo fenotipico imparziale, siamo stati in grado di identificare farmaci non previsti come NAD”, dice Ran Jing, primo autore e dottorando nel laboratorio di Duncan, che ora è un postdoctoral fellow presso la Harvard Medical School.
Per completare i loro studi, i ricercatori hanno convalidato le loro scoperte usando ratti carenti di DGUOK. Questi animali non soccombono alla malattia; tuttavia i loro fegati sono colpiti e mostrano una diminuzione della produzione di ATP. Nei modelli animali, NAD mostra una scarsa biodisponibilità, quindi i ricercatori hanno utilizzato il precursore di NAD, nicotinamide riboside (NR). Trattare ratti carenti di DGUOK che mostrano sintomi di malattia con NR ha parzialmente ripristinato i loro livelli di ATP rispetto ai ratti non trattati. Questa scoperta ha implicazioni significative per il trattamento clinico dei pazienti con MTDPS.
Per estendere queste scoperte ad altre forme di Sindrome da deplezione del DNA mitocondriale, il laboratorio di Duncan ha anche generato cellule simili alle cellule del fegato con carenza di RRM2B, la stessa mutazione che ha portato a MTDPS in Charlie Gard. Queste cellule esibivano livelli ridotti di mtDNA e ATP, anche se non drasticamente come le cellule con deficienza di DGUOK. Nonostante ciò, il trattamento con NAD ha ripristinato i livelli di ATP nelle cellule con deficienza di RRM2B, dimostrando che NAD può fornire un trattamento per più forme di MTDPS.
Il potente schermo fenotipico realizzato in questo studio ha identificato diversi farmaci che hanno aumentato la produzione di ATP. Per ripristinare ulteriormente la funzione mitocondriale, i ricercatori hanno trattato cellule carenti di DGUOK con una combinazione di NAD e trimipramina, un farmaco usato per trattare la depressione e hanno scoperto che i livelli di ATP erano ulteriormente aumentati, superando i livelli osservati nelle cellule WT. Studi di follow-up che utilizzano altre combinazioni di farmaci possono ulteriormente perfezionare il trattamento della MTDPS.
Nel complesso, questa ricerca evidenzia come la ricerca scientifica di base supporti la medicina traslazionale e clinica.
Fonte, Cell Reports