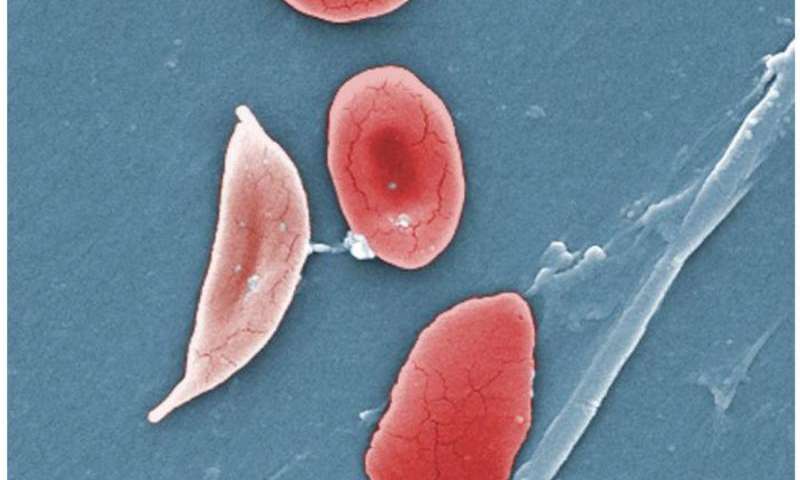
I ricercatori hanno identificato una proteina di segnalazione chiave che regola la produzione di emoglobina nei globuli rossi, un possibile bersaglio per un futuro farmaco innovativo per il trattamento dell’ anemia falciforme (SCD). Esperimenti condotti su cellule umane in coltura rivelano che il blocco della proteina riduce la caratteristica strutura a falce che distorce la forma dei globuli rossi e dà il nome alla malattia.
“Abbiamo trovato una proteina con attività specifica nei globuli rossi che potrebbe essere un bersaglio ‘druggable’, possibilmente con una piccola molecola, una pillola che i pazienti potrebbero prendere per curare l’ anemia falciforme“, ha detto il co-leader dello studio Gerd A. Blobel , scienziato presso il Children’s Hospital of Philadelphia (CHOP).
( Vedi anche:Un supplemento può alleviare il dolore dell’ anemia falciforme).
Il co-leader di Blobel e dello studio, Junwei Shi della Perelman School of Medicine presso l’Università della Pennsylvania, ha pubblicato la ricerca online il 19 luglio su Science.
La proteina di segnalazione, o chinasi chiamata HRI, è nota per regolare la produzione di emoglobina, il componente dei globuli rossi portatore di ferro. I nuovi risultati rivelano un ruolo inaspettato di HRI in un processo chiamato “commutazione dell’emoglobina”, una transizione che normalmente si verifica nei neonati durante la quale i globuli rossi passano dalla produzione di una forma fetale di emoglobina a una forma adulta. La mutazione che causa l’anemia falciforme è presente nella forma adulta di emoglobina, motivo per cui la malattia colpisce i pazienti solo dopo la nascita.
La mutazione fa sì che le cellule assumano la forma anormale della mezzaluna che intasa i vasi sanguigni e danneggia gli organi, con risultati dolorosi, a volte pericolosi per la vita. Gli ematologi sanno da tempo che i pazienti con SCD con rapporti più alti di emoglobina fetale rispetto all’emoglobina adulta hanno una forma più lieve della malattia. Il farmaco idrossiurea, che aumenta l’emoglobina fetale, è l’attuale standard di cura, ma non è efficace in tutti i pazienti. Pertanto, i ricercatori hanno cercato un trattamento migliore.
Blobel e Shi si sono affidati a uno strumento di screening utilizzando le tecniche di modifica del gene CRISPR. Shi in precedenza aveva sviluppato questo strumento per perfezionare domini funzionali specifici dei geni, senza interferire con le funzioni di interi geni. In questo particolare studio, i ricercatori si sono concentrati su una classe di domini che comprendevano protein chinasi, enzimi potenzialmente inibibili da una piccola molecola.
Lo schermo ha permesso ai ricercatori di scoprire l’HRI come la chinasi che aiuta a silenziare la produzione di emoglobina fetale nei globuli rossi adulti. Inoltre, identificando un fattore di trascrizione regolato con HRI già noto per reprimere l’emoglobina fetale, lo studio ha aggiunto un pezzo al puzzle di come l’HRI sopprima la produzione di emoglobina fetale. Quando i ricercatori hanno eliminato selettivamente la funzione di HRI, è aumentato il livello di emoglobina fetale nei globuli rossi.
Fondamentalmente, i ricercatori sono stati in grado di ridurre la falcizzazione nei globuli rossi ottenuti da pazienti con SCD, senza compromettere la vitalità o la maturazione delle cellule, suggerendo che la perdita della funzione di HRI è ben tollerata.
In esperimenti di proof-of-concept, Blobel e Shi hanno ulteriormente esaminato se un futuro farmaco che inibisce l’HRI potrebbe essere più efficace se combinato con altri farmaci progettati per aumentare l’emoglobina fetale. Gli scienziati hanno combinato la deplezione dell’HRI con il trattamento con pomalidomide, un farmaco sperimentale noto per aumentare l’emoglobina fetale . Nelle colture cellulari, l’uso della deplezione di HRI e pomalidomide insieme ha avuto un effetto più forte rispetto all’utilizzo di ciascun approccio separatamente, supportando l’idea di una terapia di combinazione per l’anemia falciforme.
“Un’altra potenziale applicazione di questo risultato”, ha aggiunto Blobel, “potrebbe essere in un’altra malattia del sangue ereditaria, la beta-talassemia, che coinvolge anche l’emoglobina anormale. Sebbene la beta-talassemia possa essere causata da molte diverse mutazioni, un sottogruppo di pazienti beta-talassemici potrebbe trarre beneficio da futuri trattamenti che hanno come bersaglio l’HRI”.
“Il nostro obiettivo a lungo termine è di condurre studi di follow-up per valutare se questo approccio migliora i risultati clinici nei pazienti”, ha affermato Blobel. “A questo punto, i nostri risultati suggeriscono che HRI è un potenziale bersaglio per un nuovo trattamento per i disturbi dell’emoglobina“.
Fonte: Science