Miopatia miofibrillare di tipo 6-immagine: Alterazione della struttura del sarcomero nel muscolo scheletrico di topi CAG-BAG3 P209L di 4 settimane. Cred: Nature Communications
La miopatia miofibrillare di tipo 6 (MFM6) è una rara malattia genetica muscolare che causa grave debolezza muscolare e una drastica riduzione dell’aspettativa di vita a causa di un’alterazione nella regolazione delle proteine muscolari. I ricercatori dell’Ospedale Universitario di Bonn (UKB) e dell’Università di Bonn hanno sviluppato un modello murino della malattia e sono stati in grado di dimostrare che un’alterazione del riciclo cellulare, tecnicamente noto come autofagia, è il principale fattore scatenante della malattia. I loro risultati sono stati pubblicati su Nature Communications.
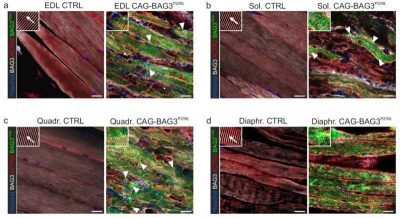
Nella miopatia miofibrillare di tipo 6 (MFM6), i sarcomeri, le unità più piccole della fibra muscolare responsabili del movimento e della tensione muscolare, si degradano. Questo processo è innescato da una proteina BAG3 difettosa (BAG3_P209L), che fa parte del complesso di autofagia selettiva associato alle chaperonine (CASA). Questo perché BAG3 svolge un ruolo chiave nell’autofagia regolata da CASA, un processo in cui le proteine danneggiate vengono eliminate o riciclate all’interno della cellula.
Gli individui affetti soffrono di debolezza muscolare a rapida progressione, danni ai nervi al di fuori del cervello e del midollo spinale e, talvolta, insufficienza cardiaca. La causa di morte più comune è l’insufficienza respiratoria dovuta alla debolezza dei muscoli scheletrici e la loro aspettativa di vita è di circa 20 anni.
Il modello murino riproduce le caratteristiche chiave della malattia
“Per riprodurre e quindi studiare al meglio le patologie del muscolo cardiaco e scheletrico osservate nei pazienti, abbiamo sviluppato un nuovo modello murino umanizzato per la MFM6. A causa di una mutazione puntiforme nel materiale genetico, BAG3 non è più in grado di svolgere la sua funzione di co-chaperone nel complesso CASA, e la sua perdita di funzione porta all’accumulo di proteine muscolari danneggiate e di conseguenza alla disgregazione dei sarcomeri“, afferma l’autore corrispondente, il Dott. Michael Hesse dell’Istituto di Fisiologia 1 dell’UKB e dell’Università di Bonn.
“Abbiamo scoperto che questi topi mostrano chiari segni di debolezza muscolare e rappresentano quindi un modello ideale per studiare il patomeccanismo di MFM6 nel muscolo scheletrico. Poiché il muscolo scheletrico è un tessuto in grado di rigenerarsi grazie alle proprie cellule staminali, è particolarmente interessante studiare le differenze tra muscolo scheletrico e muscolo cardiaco, dato che quest’ultimo è privo di cellule staminali e ha una scarsa capacità rigenerativa”.
Approcci terapeutici per migliorare la funzione muscolare
Nello studio, i ricercatori hanno osservato la degradazione dei sarcomeri, l’infiammazione e difetti nei mitocondri – le centrali energetiche delle cellule – nei muscoli scheletrici, che hanno ridotto la forza contrattile dei muscoli di circa il 90%. Inoltre, è stata osservata un’alterazione della sintesi proteica, nonché un blocco dell’autofagia e della mitofagia, un processo in cui i mitocondri vengono specificamente degradati.
Leggi anche:Cardiomiopatia ipertrofica: perché il trasporto di energia nel cuore fallisce
“Fino ad ora, non era chiaro se i difetti nei mitocondri fossero una causa o una conseguenza della malattia”, afferma l’autrice principale Kerstin Filippi. “Siamo stati in grado di dimostrare in un modello murino che gli aggregati di BAG3 e la perdita della funzione di BAG3 compromettono principalmente l’autofagia, causando così la degenerazione muscolare”.
Questo perché solo l’induzione mirata dell’autofagia mediante l’immunosoppressore rapamicina ha portato a un miglioramento significativo della funzione motoria. Anche la terapia genica nel muscolo scheletrico, che ha ridotto la quantità della proteina mutata BAG3_P209L, ha migliorato significativamente la funzione muscolare.
Astratto
“La miopatia miofibrillare 6 è una rara malattia neuromuscolare autosomica dominante causata da una sostituzione aminoacidica Pro209Leu nella co-chaperone BAG3 , che altera il turnover delle proteine muscolari e causa grave debolezza muscolare e riduzione dell’aspettativa di vita. Abbiamo generato topi transgenici che sovraesprimono la proteina BAG3 mutante umana P209L -GFP, i quali sviluppano rapidamente debolezza muscolare scheletrica, a differenza dei controlli che esprimono BAG3 WT -GFP. In questo studio dimostriamo che i topi mutanti presentano disgregazione del sarcomero, infiammazione, aggregati proteici, nuclei centralizzati e difetti mitocondriali nei muscoli scheletrici, con conseguente riduzione della forza di contrazione di circa il 90%. L’analisi omica ha rivelato un’alterazione della sintesi proteica, un blocco dell’autofagia, un’alterazione della mitofagia e la perdita di proteine del sarcomero. La modulazione delle vie metaboliche in vitro e in vivo ha mostrato che la disfunzione dell’autofagia è il principale fattore scatenante della patologia, mentre la terapia genica con silenziamento di BAG3 ha ripristinato significativamente la funzione muscolare in vivo. In sintesi, questo modello riproduce le caratteristiche principali della malattia, rivelando come gli aggregati di BAG3 e la perdita della funzione di BAG3 compromettano l’autofagia, causando la degenerazione muscolare”.
Il responsabile del gruppo di ricerca, il Prof. Dr. Bernd Fleischmann dell’Istituto di Fisiologia I dell’UKB e membro del TRA Life & Health dell’Università di Bonn, afferma: “Questo successo dimostra che abbiamo trovato un modello murino utile per testare nuovi approcci di terapia genica per curare questa devastante malattia”.
Fonte: Nature Communications