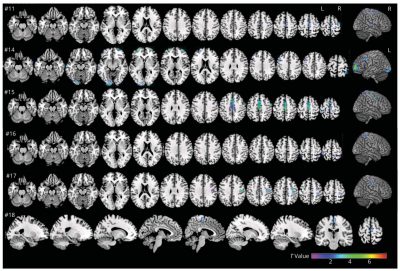
Sindrome di Lance-Adams-Immagine: la scala di colori mostra le aree con ipometabolismo del glucosio in 6 pazienti rispetto a 55 controlli sani. Crediti: Neurology
Descritto per la prima volta 60 anni fa, il mioclono cronico conseguente ad anossia cerebrale è oggi noto come sindrome di Lance-Adams. Si tratta di un grave disturbo i cui meccanismi sono stati, fino ad ora, poco compresi. Geoffroy Vellieux, Vincent Navarro e i loro colleghi del Paris Brain Institute dimostrano ora che questa condizione ha origine nella corteccia motoria. I loro risultati sono stati pubblicati sulla rivista Neurology.
La sindrome di Lance-Adams (LAS) è una grave malattia che si verifica dopo una prolungata mancanza di ossigeno al cervello, nota come anossia cerebrale, come ad esempio dopo un arresto cardiaco. Quando i pazienti riprendono conoscenza dopo un ricovero in terapia intensiva, soffrono di spasmi muscolari chiamati mioclono, che si verificano a riposo o durante il movimento. Questi sintomi interferiscono con le attività quotidiane e possono causare frequenti cadute. Soprattutto, persistono nel tempo, causando disabilità a lungo termine.
“Questa sindrome è relativamente rara e colpisce solo lo 0,5% dei pazienti che presentano mioclono dopo anossia cerebrale. Sebbene questa sindrome sia nota da tempo, i meccanismi patologici sottostanti sono rimasti un mistero“, spiega il neurofisiologo Geoffroy Vellieux, che ha completato la sua tesi di laurea in medicina sull’argomento presso il Paris Brain Institute.
La coorte più numerosa mai assemblata
Comprendere la fisiopatologia della malattia è essenziale per sviluppare approcci terapeutici efficaci. Tuttavia, nel caso della sindrome di Lance-Adams, i ricercatori hanno dovuto affrontare una sfida importante: le contrazioni muscolari dei pazienti sono molto brevi (pochi millisecondi), a volte appena percettibili, rendendone difficile l’osservazione e l’interpretazione clinica.
Inoltre, sebbene l’elettroencefalografia (EEG) fosse già stata utilizzata dai medici nei primi studi sulla LAS negli anni ’60, essa non consente un’identificazione precisa dell’origine delle anomalie cerebrali.
Per far luce su queste incertezze, i ricercatori del team “Epilessia clinica e sperimentale”, co-diretti dal Prof. Navarro (AP-HP, Università della Sorbona), hanno studiato 18 pazienti curati presso l’ospedale Pitié-Salpêtrière: una coorte significativa per una malattia rara e la più grande mai assemblata per questa condizione.
Grazie ad approfonditi esami neurofisiologici e neuroradiologici (tra cui elettromiografia, elettroencefalografia e tomografia a emissione di positroni), il team ha dimostrato che il mioclono dei pazienti ha origine prevalentemente nella corteccia cerebrale, in particolare nella corteccia motoria.
“Questi risultati confermano l’intuizione iniziale di Raymond Adams e James Lance, i neurologi che nel 1963 diedero il nome alla malattia“, osserva Navarro.
Nuove vie terapeutiche
All’interno della coorte, 11 pazienti hanno manifestato anche crisi epilettiche. Sorprendentemente, per 8 di loro, il mioclono si è ridotto significativamente in seguito a queste crisi. “Sulla base di questa osservazione, abbiamo proposto la terapia elettroconvulsivante a una paziente, che prevede la somministrazione di uno stimolo elettrico attraverso il cervello, per riprodurre le crisi in modo controllato. Il trattamento ha alleviato significativamente i suoi sintomi, che erano resistenti ai farmaci“, aggiunge il ricercatore.
Questa scoperta apre la strada a nuove strategie per modulare l’attività corticale anomala.
Infine, i ricercatori intendono condurre nuovi studi per individuare esattamente quali popolazioni e reti di neuroni, all’interno della corteccia motoria, innescano la cascata di segnali che causa il mioclono, con l’obiettivo di sviluppare nuovi trattamenti, farmacologici o di altro tipo, per migliorare la qualità della vita dei pazienti.
Fonte:Neurology