Immagine: Public Domain.
Riepilogo: i ricercatori hanno identificato come specifiche mutazioni genetiche causino la SLA. Secondo loro, il percorso potrebbe anche essere responsabile dello sviluppo della demenza frontotemporale.
I ricercatori della University of Maryland School of Medicine (UMSOM) hanno identificato il modo in cui alcune mutazioni genetiche causano la sclerosi laterale amiotrofica (SLA). Il percorso identificato dai ricercatori può anche essere responsabile di una certa forma di demenza correlata alla SLA. La scoperta potrebbe offrire potenziali nuovi approcci per il trattamento di questa condizione devastante, che provoca una paralisi progressiva, fatale e talvolta un deterioramento mentale simile alla malattia di Alzheimer.
La loro scoperta è stata pubblicata questa settimana negli Atti della National Academy of Sciences (PNAS) ed è stata realizzata da ricercatori dell’Università di Harvard, dell’Università di Auckland, del King’s College di Londra e della Northwestern University.
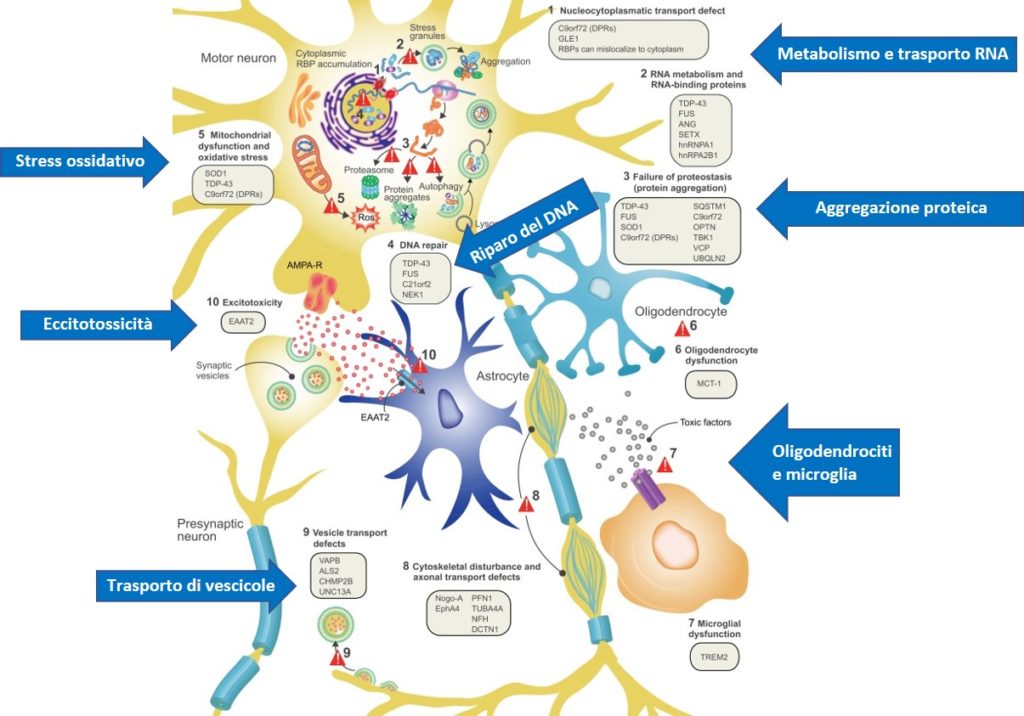
Oltre 5.000 americani vengono diagnosticati con SLA ogni anno, una condizione che di solito è fatale e non ha cura. I pazienti con la SLA perdono lentamente la capacità di muovere i muscoli con conseguenti problemi con le funzioni di base come la respirazione e la deglutizione. Circa la metà dei pazienti con SLA sviluppa anche demenza. Studi genetici su famiglie con una predisposizione allo sviluppo della SLA hanno dimostrato che la condizione può essere associata a determinate mutazioni genetiche. Alcune di queste mutazioni coinvolgono il gene UBQLN2 che regola lo smaltimento di “rifiuti” in particolare proteine ripiegate male dalle cellule del corpo. Fino ad ora, i ricercatori non hanno compreso appieno come le mutazioni UBQLN2 interferiscono con questo percorso e causino la SLA.
“Abbiamo mappato il processo mediante il quale le mutazioni del gene ubiquilin-2 (UBQLN2) interrompono un importante percorso di riciclaggio che le cellule usano per sbarazzarsi dei loro rifiuti”, ha dichiarato Mervyn Monteiro, PhD, Professore di Anatomia e Neurobiologia, affiliato all’UMSOM Centro di ingegneria e tecnologia biomedica (BioMET) presso UMSOM. “Senza questo riciclaggio, le proteine mal ripiegate si accumulano nella cellula nervosa e diventano tossiche, alla fine distruggendo la cellula. Questa distruzione potrebbe portare a disturbi neurodegenerativi come la SLA“.
Per studiare in che modo le mutazioni di UBQLN2 causano la SLA, il gruppo del Dr. Monteiro ha utilizzato sia le cellule umane sia i modelli di topo mutante UBQLN2 per le indagini. I modelli di topo, che hanno descritto in una pubblicazione PNAS del 2016, imitano la progressione della malattia nelle persone che ereditano queste mutazioni genetiche.
Il gruppo del Dr. Monteiro ha rimosso per la prima volta il gene UBQLN2 dalle cellule umane e ha scoperto che questa operazione bloccava completamente il percorso di riciclaggio. I ricercatori hanno quindi reintrodotto nelle cellule il gene normale o una delle cinque mutazioni genetiche. Hanno scoperto che la reintroduzione del normale UBQLN2 ha ripristinato il percorso di riciclaggio mentre tutte e cinque le mutazioni genetiche non sono riuscite a riavviare il percorso.
Vedi anche: Metaboliti plasmatici utili sia per la diagnosi che per monitorare la progressione della SLA
Utilizzando il modello murino, il Dott. Monteiro e i suoi colleghi hanno delineato il motivo dell’interruzione del percorso in presenza di mutazioni genetiche. Hanno scoperto che i topi con le mutazioni genetiche avevano livelli ridotti di una certa proteina chiamata ATP6v1g1, che è una parte essenziale di una pompa che acidifica il contenitore di rifiuti cellulari al fine di avviare il processo di scomposizione e riciclaggio.
“I nostri nuovi risultati sono entusiasmanti perché simili difetti di acidificazione sono stati riscontrati nell’Alzheimer, nel Parkinson e nella sindrome di Down”, ha affermato il Dott. Monteiro. “Ciò suggerisce che il ripristino del difetto potrebbe avere ampie implicazioni non solo per il trattamento della SLA, ma probabilmente anche per altre malattie neurodegenerative”.
Lo studio di ricerca è stato supportato da sovvenzioni del Centro Packard per la ricerca sulla SLA presso la Johns Hopkins University e dalla SLA Association e il National Institutes of Health (numero di sovvenzione: R01-NS100008).
Fonte: Neurosciencenews