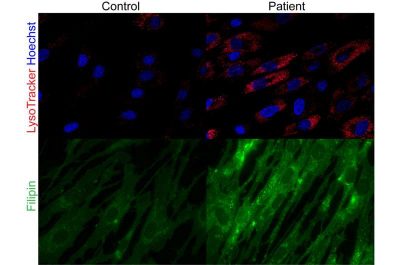
Malattia multiorgano-Immagine: fibroblasti cutanei in un individuo sano (a sinistra) e fibroblasti cutanei con lisosomi ingrossati e accumulo di colesterolo in un paziente con mutazioni SPNS1 (a destra). Crediti: Duke-NUS Medical School
Un team di scienziati guidato dalla Duke-NUS Medical School ha risolto il mistero di una rara malattia mai diagnosticata prima che colpisce più organi, gettando nuova luce sulla sua causa e offrendo nuove speranze di cura.
Pubblicato sul Journal of Clinical Investigation, lo studio ha individuato le mutazioni in un gene chiamato SPNS1 come causa sottostante del disturbo, che influisce sul modo in cui le cellule riciclano le molecole di grasso.
I ricercatori hanno scoperto che “versioni difettose di questo gene interrompono la funzione dei lisosomi (il sistema di riciclaggio cellulare del corpo), provocando un accumulo dannoso di grassi e colesterolo e, infine, danni progressivi al fegato e ai muscoli”.
La nuova condizione fa parte della famiglia delle malattie da accumulo lisosomiale, un gruppo di oltre 70 malattie rare causate da un’interruzione del riciclaggio cellulare.
La scoperta è nata dallo studio di due famiglie non imparentate, i cui figli presentavano una malattia epatica inspiegabile, debolezza muscolare e altri sintomi. L’analisi genetica ha rivelato mutazioni in entrambe le copie di SPNS1, un trasportatore cruciale per il trasporto delle molecole di grasso scomposte dal lisosoma al resto della cellula, dove vengono riciclate.
La ricerca si basa su un precedente studio condotto dalla Duke-NUS che aveva individuato il ruolo del gene SPNS1 nel riciclaggio dei grassi scomposti.
La Dott.ssa He Menglan, studentessa di medicina e dottorato presso la Duke-NUS e prima autrice dello studio, ha affermato che i risultati rappresentano un tassello fondamentale per comprendere una malattia rimasta a lungo un mistero: un tipo importante di grasso che i nostri sistemi di riciclo cellulare elaborano sono i fosfolipidi, che sono i mattoni fondamentali delle membrane cellulari. Negli individui sani, SPNS1 sposta i fosfolipidi degradati fuori dai lisosomi per essere riutilizzati per riparare le membrane o convertiti in energia di riserva per l’organismo.
“Quando questo complesso processo fallisce nei pazienti con mutazioni SPNS1, il riciclaggio dei grassi viene interrotto, causando danni ai tessuti, in particolare nei muscoli e nel fegato”, spiega la Dr.ssa He Menglan.
I ricercatori hanno scoperto che questi problemi peggioravano quando un sistema chiave di rilevamento dei nutrienti veniva interrotto, evidenziando l’importanza di SPNS1 nell’aiutare le cellule a rispondere allo stress dei nutrienti e a mantenere l’equilibrio energetico.
Il Professor David Silver, viceDirettore del programma di malattie cardiovascolari e metaboliche della Duke-NUS e autore principale dello studio, ha affermato: “SPNS1 è presente in ogni cellula umana e svolge un ruolo chiave nel riciclo dei fosfolipidi. I nostri studi hanno rivelato che il riciclo dei fosfolipidi da parte dei lisosomi svolge un ruolo cruciale nel regolare il modo in cui le cellule mantengono livelli normali di altri lipidi importanti come grassi e colesterolo. Questi risultati aprono opportunità per esplorare l’importanza di SPNS1 in altre malattie come il cancro“.
Grazie a queste intuizioni, il team sta collaborando con N=1 Collaborative, un’organizzazione che sviluppa terapie personalizzate per malattie estremamente rare, per tradurre le proprie scoperte in soluzioni pratiche.
La Dott.ssa Marlen Lauffer, ricercatrice senior presso il Dutch Center for RNA Therapeutics del Leiden University Medical Center e coautrice dello studio, ha sottolineato l’importanza di applicare questi risultati alla cura dei pazienti. “Sfruttando quanto appreso da questa ricerca, stiamo collaborando con l’N = 1 Collaborative per creare un trattamento su misura per i bambini del nostro studio affetti da questa condizione. Questo lavoro include l’esplorazione di modi per correggere il trasporto difettoso del grasso utilizzando nuove terapie genetiche. Il nostro obiettivo è trasformare le conoscenze scientifiche in terapie che migliorino la qualità della vita e diano speranza ad altre famiglie che affrontano sfide simili”.
Il Dott. Lauffer ha aggiunto che comprendere la causa precisa della malattia consente ai ricercatori di progettare trattamenti che agiscono direttamente sui percorsi interrotti, offrendo opzioni ai pazienti che attualmente non hanno a disposizione alcun percorso terapeutico.
La signora Dalila Sabaredzovic, madre di due dei bambini coinvolti nello studio, spera che questa scoperta rappresenti il primo passo verso il miglioramento della qualità della vita dei suoi figli e di altre persone affette dalla stessa patologia.
“Sono davvero grata che ora abbiamo una base su cui poggiare e che il lavoro stia progredendo verso l’esplorazione di percorsi terapeutici. Ci sentiamo rafforzati in molti modi in cui prima non ci sentivamo e speriamo davvero che questa ricerca possa stimolare non solo la comprensione del gene SPNS1 e della patologia che causa, ma anche la strada verso una cura”, ha affermato.
Leggi anche:Disturbi da accumulo lisosomiale: gli scienziati invertono gli effetti
Spiegano gli autori:
“SPNS1 è un trasportatore lisosomiale che media il recupero dei lisoglicerofosfolipidi, i prodotti di degradazione del catabolismo dei fosfolipidi lisosomiali. Tuttavia, il ruolo del trasporto e del recupero dei lisolipidi nella regolazione dell’omeostasi lipidica cellulare e nella patologia è ancora poco noto. In questo studio, abbiamo identificato due famiglie con varianti bialleliche di SPNS1 con perdita di funzione che si presentavano principalmente con danno progressivo al fegato e al muscolo striato. I fibroblasti dei pazienti accumulavano lisofosfolipidi, inclusi lisoplasmalogeni e colesterolo, nei lisosomi con plasmalogeni cellulari ridotti. In particolare, la carenza di SPNS1 ha portato a una ridotta biogenesi di goccioline lipidiche citosoliche contenenti trigliceridi ed esteri del colesterolo. Dal punto di vista meccanico, abbiamo scoperto che i lisofosfolipidi trasportati da SPNS1 nel citosol contribuivano quantitativamente alla sintesi dei trigliceridi, mentre l’accumulo lisosomiale di liso-etere-fosfolipide inibiva l’uscita lisosomiale del colesterolo, effetti che risultavano potenziati dall’inibizione di mTOR. Questi risultati supportano un’associazione gene-malattia e rivelano una connettività tra il trasporto lisosomiale dei lisofosfolipidi e l’accumulo di energia cellulare di riserva sotto forma di trigliceridi, nonché nella regolazione dell’omeostasi del colesterolo, processi che diventano importanti in condizioni di limitazione nutrizionale“.
Astratto grafico

Il Professor Patrick Tan, vicePreside senior per la ricerca presso la Duke-NUS, ha affermato: “Questi risultati dimostrano il potere della medicina di precisione. Collegando sintomi insoliti dei pazienti a specifiche mutazioni genetiche, i ricercatori scoprono nuovi percorsi patologici e sviluppano trattamenti mirati. Questo approccio non solo fornisce risposte alle famiglie colpite da malattie rare, ma apre anche le porte a progressi medici più ampi. Questa scoperta ci ricorda che anche le condizioni più rare e misteriose possono essere risolte, quando scienziati, medici e famiglie lavorano insieme“.