SLA-Immagine Credito: Cell Genomics (2025).
Ricercatori della Facoltà di Medicina Temerty dell’Università di Toronto hanno scoperto diffuse alterazioni genetiche e proteiche nelle cellule cerebrali delle persone affette da sclerosi laterale amiotrofica (SLA), che potrebbero contribuire a far luce sui meccanismi della malattia.
Lo studio, pubblicato di recente su Cell Genomics, descrive anche lo sviluppo di uno strumento di intelligenza artificiale che utilizza l’apprendimento profondo per aiutare a prevedere se i cambiamenti che contribuiscono alla SLA e ad altre malattie neurodegenerative, siano presenti nelle cellule.
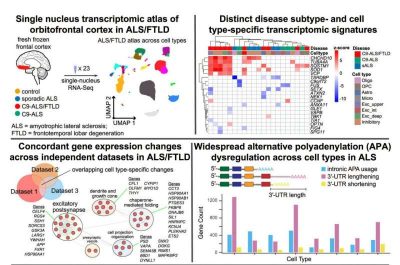
Paul McKeever, coautore principale dello studio, afferma che i ricercatori hanno sviluppato un “atlante” della disregolazione dell’RNA nelle cellule cerebrali della corteccia orbito-frontale attraverso un’ampia analisi dei cambiamenti genetici. Questa regione cerebrale è associata a cambiamenti comportamentali nella SLA e nella degenerazione lobare frontotemporale (FTLD).
“Molti ricercatori studiano la disregolazione di specifiche proteine leganti l’RNA nelle cellule cerebrali della SLA e il modo in cui ciò influisce sulla funzione cellulare”, afferma McKeever, ricercatore post-dottorato con la professoressa Janice Robertson, ricercatrice presso il Tanz Center for Research in Neurodegenerative Diseases e titolare della cattedra James Hunter Family Chair in ALS Research. “Abbiamo fatto un passo indietro e ci siamo chiesti se fosse possibile identificare quelle proteine nelle cellule, come potrebbero influenzare l’elaborazione dell’RNA e, di conseguenza, come i geni vengono espressi in queste cellule”.
Metodi di studio e approccio alla ricerca
“Il nostro approccio è stato ampio e profondamente comparativo, consentendoci di esaminare molti tipi di cellule e sottotipi di malattie uno accanto all’altro e di scoprire modelli di cambiamento mai osservati prima”.
Il team ha utilizzato una tecnica chiamata sequenziamento dell’RNA a singolo nucleo, che analizza l’RNA proveniente da singoli nuclei cellulari anziché da cellule intere, ed è ideale per il sequenziamento di campioni di pazienti precedentemente congelati. La comprensione delle sequenze di RNA fornisce informazioni sui percorsi cellulari interessati dalla malattia e sui meccanismi coinvolti, e può portare all’identificazione di nuovi bersagli terapeutici.
McKeever, in collaborazione con i ricercatori del Tanz Center e del dipartimento di medicina di laboratorio e patobiologia della Temerty Medicine, ha sequenziato l’RNA da campioni di tessuto cerebrale delle dimensioni di un pisello contenenti diversi tipi di cellule provenienti da persone affette da forme genetiche e sporadiche di SLA, nonché da persone senza SLA.
Risultati chiave e implicazioni
I loro risultati mostrano alcune differenze nella trascrizione dell’RNA tra tipi di cellule, regioni cerebrali e sottotipi di SLA, ma anche alcuni modelli coerenti di cambiamenti tra i tipi di cellule, in particolare nei neuroni, suggerendo che tali cambiamenti sono meccanismi fondamentali che guidano l’avanzamento della malattia.
“Abbiamo scoperto che esistono cambiamenti condivisi e distinti sia tra i tipi cellulari che tra i tipi di cellule malate, e questo si è esteso a studi indipendenti che hanno realmente convalidato i nostri risultati”, afferma McKeever. “Questo fornisce una comprensione più dettagliata delle differenze tra le cellule cerebrali nella SLA e nella FTLD e illumina i meccanismi della malattia specifici dei tipi cellulari, offrendo una risorsa preziosa per il progresso della ricerca terapeutica“.
McKeever afferma che una migliore comprensione delle alterazioni dell’RNA e delle reti proteiche cellulari può aiutare a identificare le firme genetiche, o biomarcatori, di una malattia, utili per la diagnosi. Potrebbe anche aiutare i ricercatori a comprendere meglio i percorsi cellulari che portano alla malattia.
Uno dei cambiamenti più diffusi osservati dai ricercatori in molte cellule cerebrali della SLA, in particolare nei neuroni e nella glia, è stata la disregolazione della poliadenilazione alternativa (APA), un processo che avviene dopo la trascrizione dell’RNA e che influenza il processo di traduzione in proteine.
Sviluppo dello strumento di deep learning
Sulla base di questa scoperta, Gary Bader, Professore di genetica molecolare presso il Donnelly Center for Cellular and Biomolecular Research e il suo dottorando dell’epoca, Aiden Sababi, hanno utilizzato la biologia computazionale per sviluppare uno strumento di apprendimento profondo predittivo .
Hanno utilizzato i dati dell’analisi del sequenziamento dell’RNA di McKeever per prevedere se una cellula avrebbe avuto un’APA disregolata in base alle sue sequenze di RNA e alle reti di proteine leganti l’RNA.
Sababi, ora ricercatore post-dottorato presso la McGill University, afferma che lo strumento di apprendimento profondo è prezioso per prevedere quali cellule potrebbero avere APA disregolati, ma anche per generare idee per la ricerca futura sulle proteine che hanno maggiori probabilità di guidare lo sviluppo e la progressione delle malattie.
“Cerchiamo di concentrarci sulla funzione biologica, piuttosto che sulla sola previsione. Non vogliamo solo vedere dove cambia l’APA, ma anche capire perché cambia nella SLA, quindi lo strumento è prezioso per formulare ipotesi e aprire nuove strade alla ricerca”, afferma Sababi.
“Il vantaggio di questo strumento di deep learning è che, una volta che il modello fornisce previsioni affidabili, è possibile iniziare a capire perché il modello le fornisce. Il modello ha confermato le prove del team del Tanz Center secondo cui la disregolazione dell’APA è in atto, e ora la domanda è perché“.
Leggi anche: SLA: farmaco candidato rallenta la progressione e preserva la funzione muscolare
Direzioni future
Graham Collingridge, Direttore del Tanz Center, afferma che la ricerca rappresenta un importante passo avanti nella comprensione dello sviluppo della malattia.
“La SLA è una malattia devastante e comprenderne le cause e sviluppare trattamenti efficaci è una priorità assoluta per i ricercatori di Tanz e altri in tutto il mondo“, afferma. “Questo studio e lo strumento di apprendimento profondo associato che è stato sviluppato costituiscono passi importanti in questa direzione”.
Sababi ha utilizzato un set di dati relativamente piccolo per addestrare il modello di apprendimento profondo, ma sulla base dei risultati promettenti di questo studio, il team di ricerca desidera ampliarne le capacità.
Robertson, McKeever, Sababi e Bader hanno recentemente ricevuto un McLaughlin Accelerator Grant in Genomic Medicine dall’Università di Toronto per sviluppare il modello, ampliarne le capacità e potenzialmente utilizzarlo per analizzare dati simili su altre malattie neurodegenerative e tumori.
“Questa nuova sovvenzione ci consentirà di far progredire il modello, di addestrarlo su set di dati più ampi e, auspicabilmente, di convalidare ciò che vediamo nei dati attuali”, afferma Sababi. “Man mano che acquisiamo maggiore sicurezza con questo modello, possiamo formulare ipotesi migliori sui meccanismi delle malattie e convalidare meglio gli approcci che abbiamo adottato. Credo che ci stiamo avvicinando ad un impatto positivo da questo ambito della biologia“.
Fonte: Cell Genomics