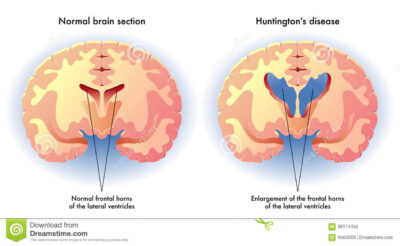
Malattia di Huntington/Immagine credit public domain.
La malattia di Huntington ha a lungo sfidato i tentativi di salvare i neuroni sofferenti. Un nuovo studio pubblicato su Cell Reports dimostra che il trapianto di cellule progenitrici gliali umane sane nel cervello di modelli animali adulti della malattia non solo ha rallentato il declino motorio e cognitivo, ma ha anche prolungato la durata della vita. Questi risultati ampliano la nostra comprensione della patologia di Huntington e aprono una potenziale strada alle terapie cellulari negli adulti che già presentano sintomi.
La glia è una custode essenziale dei neuroni. Il ripristino di un sano supporto gliale, anche dopo la comparsa dei sintomi della malattia di Huntington, potrebbe ripristinare l’espressione genica neuronale, stabilizzare la funzione sinaptica e ritardare significativamente la progressione della malattia.
“Questo studio sposta la prospettiva sulla malattia di Huntington da una visione neurocentrica a una che mostra un ruolo cruciale della patologia gliale nel determinare la disfunzione sinaptica. Ci dice anche che il cervello adulto ha ancora la capacità di ripararsi quando si prendono di mira le cellule giuste”, dice Steve Goldman, MD, PhD, co-Direttore del Centro per la neuromedicina traslazionale dell’Università di Rochester e autore principale dello studio.
Malattia di Huntington: oltre i neuroni
La malattia di Huntington è una malattia cerebrale ereditaria causata da una mutazione nel gene huntingtina. Questa mutazione porta alla produzione di una proteina anomala che danneggia gradualmente le cellule nervose, in particolare in una regione chiamata striato, causando problemi di movimento, sbalzi d’umore e declino cognitivo.
L’approccio scientifico a questa malattia si è tradizionalmente concentrato sul salvataggio o sulla sostituzione dei neuroni colpiti, ma decenni di ricerca nel laboratorio Goldman hanno dimostrato che le cellule di supporto del cervello, chiamate glia, svolgono un ruolo cruciale nello sviluppo della malattia.
Un tempo considerate una semplice “colla” che tiene fermi i neuroni, ora si sa che la glia regola la salute neuronale, controlla l’infiammazione e mantiene l’equilibrio chimico del cervello. Nella malattia di Huntington, la glia diventa disfunzionale e può contribuire al danno neuronale. Sostituendo la glia malata con quella sana, gli scienziati sperano di ripristinare l’ambiente di supporto di cui i neuroni hanno bisogno per funzionare correttamente, potenzialmente preservando le cellule nervose perse a causa della malattia.
Trapianto di glia sana in topi sintomatici
I ricercatori hanno utilizzato topi R6/2, un modello consolidato di malattia di Huntington che sviluppa sintomi motori e cognitivi simili a quelli osservati negli esseri umani. A cinque settimane di età, quando i sintomi sono appena iniziati ma prima di un grave declino, questi topi hanno ricevuto iniezioni direttamente nei loro striati di cellule progenitrici gliali umane, glia in fase iniziale che può maturare in diversi tipi di cellule gliali . I topi sono stati testati su compiti che misuravano coordinazione, movimento, memoria e ansia.
Il team ha utilizzato il sequenziamento dell’RNA a singolo nucleo per vedere quali geni fossero attivati o disattivati nei neuroni dei topi trattati. Hanno anche etichettato singoli neuroni con un virus della rabbia modificato per visualizzarne le ramificazioni (dendriti) e i punti di connessione (spine).
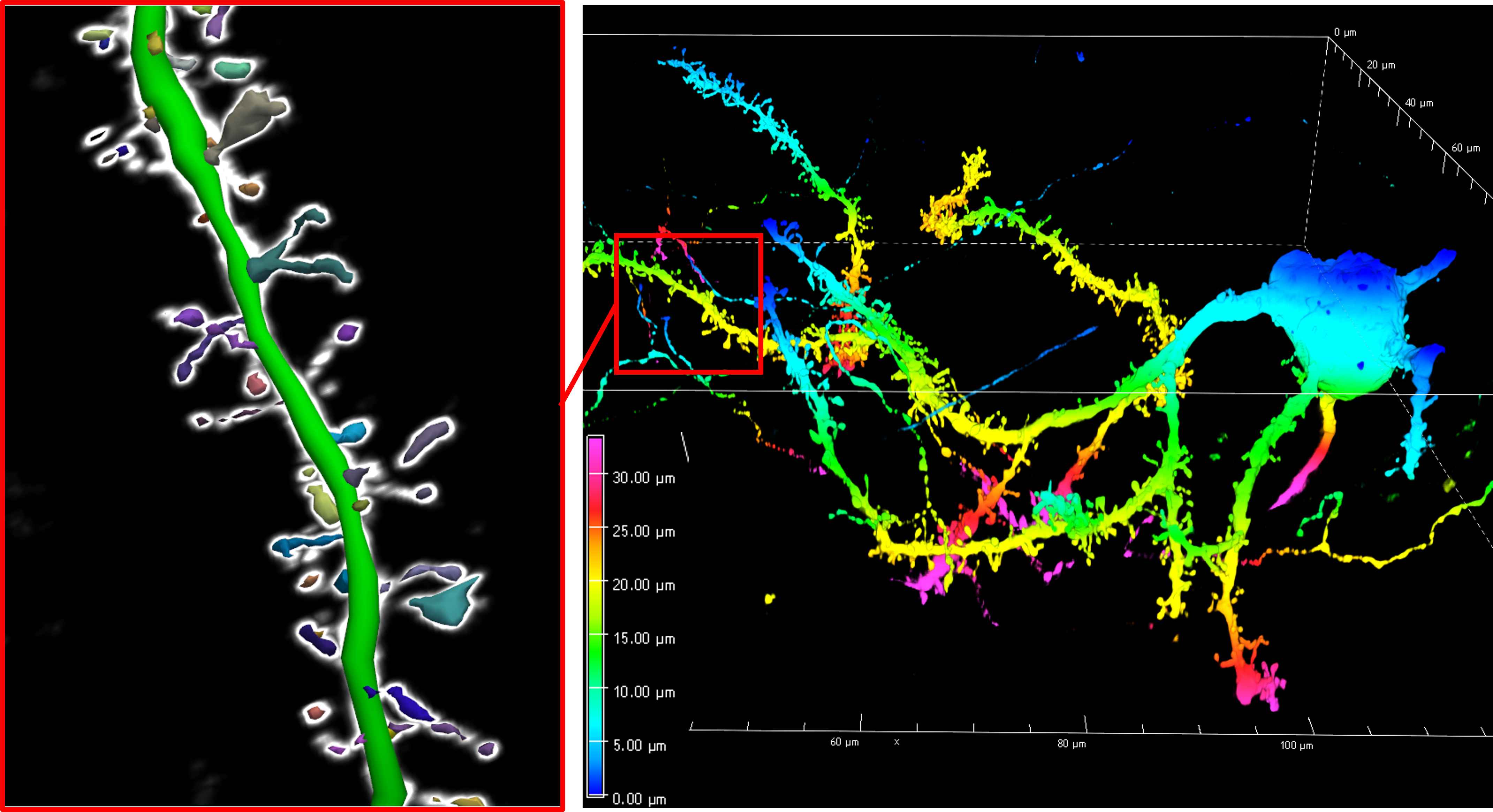
Dai miglioramenti motori al ripristino sinaptico
I topi trattati hanno mostrato un chiaro ritardo nel deterioramento motorio e cognitivo e sono vissuti diverse settimane in più rispetto ai topi HD non trattati.
I neuroni nei topi R6/2 normalmente perdono l’espressione dei geni coinvolti nel mantenimento delle sinapsi funzionali, le connessioni tra le cellule nervose. Dopo il trapianto di glia, molti di questi geni sono stati effettivamente riattivati. Inoltre, mentre i neuroni nei topi che modellano la malattia di Huntington presentano tipicamente meno rami e spine, nei topi Huntington a cui è stata data una glia sana, la ramificazione dendritica e la densità delle spine sono tornate a livelli prossimi alla normalità.
“Sebbene il trattamento sia iniziato dopo la comparsa dei sintomi, si sono comunque osservati miglioramenti significativi, evidenziando il potenziale di un intervento sugli adulti”, ha affermato il coautore dello studio Abdellatif Benraiss, PhD, dell’University of Rochester Medical Center. “Questo studio dimostra che concentrarsi sulla salute delle cellule gliali, in questo caso trapiantando cellule sane, può avere un impatto significativo sulla progressione della malattia, non solo nei modelli neonatali, ma anche negli adulti che presentano già sintomi”.
Leggi anche:Malattia di Huntington: svelata la prima immagine dettagliata delle fibrille
Espansione e perfezionamento delle strategie basate sulle cellule
I ricercatori ritengono che il trapianto di cellule di supporto sane possa diventare parte di una strategia di trattamento multidisciplinare, da solo o in associazione a un approccio di targeting genico. La ricerca futura dovrà determinare la somministrazione, il dosaggio e la tempistica ottimali per questa strategia. Oltre a ciò, tuttavia, la combinazione della sostituzione gliale con altre terapie, come la riduzione dell’espressione di huntingtina mutata e la sostituzione dei neuroni persi, potrebbe produrre benefici ancora maggiori.
“Sebbene i modelli murini non riassumano tutti gli aspetti della malattia di Huntington umana, queste scoperte ampliano il panorama terapeutico, includendo la sostituzione o la riparazione delle cellule gliali come una potenziale strategia di trattamento interessante“, ha affermato Goldman.
Tra gli altri coautori figurano i co-primi autori Carlos Villanueva e Nguyen Huynh, nonché John N. Mariani, Benjamin Mansky, Ashley Tate, Signe Syshoj Lorenzen e Devin Chandler-Militello del Center for Translational Neuromedicine, che comprende laboratori dell’Università di Rochester e dell’Università di Copenaghen. La ricerca è stata finanziata dalla Fondazione Lundbeck, dalla Fondazione Novo Nordisk, da Sana Biotechnology, dal National Institute on Aging, dalla Fondazione CHDI e dall’Huntington’s Disease Golf Classic.